All published articles of this journal are available on ScienceDirect.

Hematological Disorders in Patients with Systemic Lupus Erythematosus
Abstract
This article is a review of different management strategies for the hematological manifestations of systemic lupus erythematosus (SLE), the strategies include immunosuppressive drugs, some noval therapies and B-cell depletion for refractory thrombocytopenia in patients with SLE and in antiphospholipid antibody syndrome associated with SLE. The researcher questions the validity of the current classic treatment modes and the article explores the relationships between SLE hematological manifestations and the level of morbidity and mortality burden and focuses on the pathophysiology, diagnostic approaches and management strategies of these manifestations.
The researcher focuses on hematological abnormalities because they are the commonest among most manifestations in SLE seen in Anemia, leucopenias and thrombocytopenia. They commonly result from an immune mediated bone marrow failure, excessive peripheral cells destruction or certain drugs and infections. There is also an association between anti-phospholipid antibody syndrome (APS) and SLE referred to as secondary APS or SLE-APS. Furthermore, it was recently found that mycophenolatemofetil acts as corticosteroids and as cyclophosphamide sparing agent. Although there is no specific therapy for cytopenias in SLE, corticosteroids remain the mainstay in the treatment of these patients along with less used other conventional treatment options such as azathioprine, cyclophosphamide and human normal immunoglobulin. There are other novel therapies such as thrombopoietin receptor agonists in thrombocytopenia and the use of autologous hematopoitic stem cells transplantation in refractory SLE-APS that are under review. Some of these therapies include thrombopoietin receptor agonists in thrombocytopenia and the use of autologous hematopoitic stem cells transplantation in refractory SLE-APS.
The study concludes that treatment of hematological abnormalities is challenging because the treatment itself can cause undue complications sometimes such as granulocytosis due to infection or the use of high doses of steroids and may occur during acute exacerbations of SLE. It is important to take these factors into consideration for disease therapy and management.
Publication Abstract:
This article is a review of different management strategies for the hematological manifestations of systemic lupus erythematosus (SLE). The strategies include immunosuppressive drugs, some novel therapies and B-cell depletion for refractory thrombocytopenia in patients with SLE and in anti-phospholipid antibody syndrome associated with SLE. The researcher questions the validity of the current classic treatment modes and the article explores the relationships between SLE hematological manifestations and the level of morbidity and mortality burden while it focuses on the pathophysiology, diagnostic approaches and management strategies. The study concludes that hematological abnormalities are the commonest among most manifestations in SLE, and that their treatment is challenging because the treatment itself can cause undue complications sometimes such as granulocytosis due to infection or the use of high doses of steroids and may occur during acute exacerbations of SLE. It is important to take these factors into consideration for disease therapy and management.
INTRODUCTION
This article investigates the relationships between SLE hematological manifestations and the levels of morbidity and mortality burden from these manifestations. The frequency of hematological disorders in SLE are the highest among all other manifestations in Saudi Arabia [1]. Hematological disorders were reported in 82.7% of patients with SLE in a study conducted in Saudi Arabia [1]. The major manifestations are anemia, leucopenia, thrombocytopenia and antiphospholipid syndrome (APS). One study of 126 patients with SLE showed that 47% had neutropenia, 27% had thrombocytopenia, 20% with lymphopenia and 13% had hemolytic anemia [2,3]. These vary widely among patients, and they are listed as the most common manifestations of SLE in the American College of Rheumatology (ACR) criteria for SLE classification. This includes hemolytic anemia with reticulocytosis, leucopenia (<4.0x10⁹/L) or lymphopenia (<1.5x10⁹/L) on two or more occasions, or thrombocytopenia (<100x10⁹/L) in the absence of offending drugs [4,5].
Furthermore, antiphospholipid antibodies are found in approximately 30-40% of patients with SLE, but only about 10% have APS. Approximately half of APS cases are not associated with another rheumatic disease. In a study of 100 patients with confirmed venous thrombosis and no history of SLE, anti cardiolipin (aCL) antibodies were found in 24% and lupus anticoagulants (LA) in 4% [6,7].
This article seeks to map the pathophysiology, diagnostic approaches and management strategies of different ematological manifestations in SLE patients. It takes into account Anemia, Leucopenia, Neutropenia, Thrombocytopenia, Pancytopenia, Leukocytosis, Lymphadenopathy and Splenomegaly. After defining the condition, the classic therapy is described then some management strategies that shift from the classic modes of treatment are suggested.
Anemia
It is a common hematological abnormality in SLE that is defined as hemoglobin levels of < 12g/dL for women and <13.5 g/dL for men. It is categorized into the following: anemia of chronic disease (ACD), which is the most common form (60%-80%), iron deficiency anemia (IDA), autoimmune hemolytic anemia (AIHA), and anemia due to chronic renal insufficiency. In a cohort comprising 132 anemic patients with SLE, ACD was found in 37.1% of the cases, IDA in 35.%, AIHA in 14.4% and other causes of anemia in 12.9% of the patients [8].
ACD results from suppressed erythropoiesis secondary to chronic inflammation (normocytic and normochromic, with a relatively low reticulocyte count, low to normal serum iron, adequate bone marrow iron stores and elevated serum ferritin level) [9]. Low levels of erythropoietin due to chronic inflammation or renal insufficiency and presence of anti-erythropoietin autoantibodies which are associated with European Consensus Lupus Activity Measurement (ECLAM) high score are found in some patients [8,10].
IDA is defined by serum ferritin below 20 μg/dl [8]; it is common and may be the result of menorrhagia or increased gastrointestinal blood loss because of long term use of corticosteroids [8,9].
AIHA is characterized by elevated reticulocyte counts, low haptoglobin levels, increased indirect bilirubin concentration and a positive direct Coombs' test [9]. It has been noted in up to 10% of patients with SLE. The presence of hemolytic anemia may associate with manifestations of severe disease such as renal disease, seizures and serositis. The presence of both immunoglobulin and complements on red blood cells is usually associated with some degree of hemolysis, while presence of complements alone (C3 and /or C4) is often not associated with hemolysis [9].
Other types of anemia in SLE are rare. Pure red cell aplasia (PRCA), pernicious anemia (PA), and aplastic anemia have been reported in these patients. PRCA is characterized by antibodies directed against either erythropoietin or bone marrow erythroblasts, aplastic anemia is mediated by auto antibodies against bone marrow precursors [8,9].
Furthermore, drugs, myelofibrosis, sideroblastic anemia, hemophagocytic syndrome and thrombotic microangiopathy are also implicated in the causation of SLE anemia (Fig. 1).
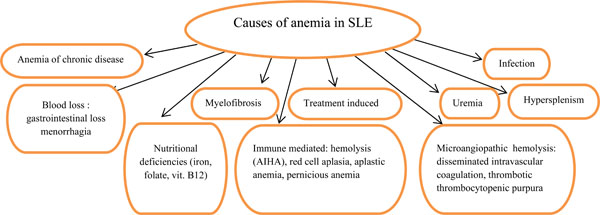
Causes of anemia in SLE.
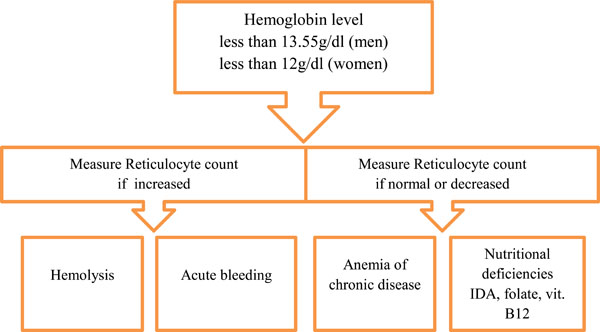
Initial approach to Anemia in SLE.
Microangiopathic hemolytic anemia (MAHA) is manifested by schistocytes on peripheral blood smear, elevated serum levels of lactate dehydrogenase and bilirubin. Many affected patients also have thrombocytopenia, renal involvement, fever and neurologic symptoms. These features are compatible with a diagnosis of thrombotic thrombocytopenic purpura (TTP). However, the pathogenesis of TTP in these patients is likely heterogeneous, as it may reflect vasculitis or antiphospholipid syndrome as well [9].
Leucopenia
It is a typical feature of SLE which may occur as a result of lymphopenia, neutropenia or a combination of both. The prevalence of lymphopenia in SLE ranges from 20 to 81% and its degree may correlate with disease activity. Both T and B lymphocytes are reduced, while natural killer cells are typically increased [9]. Reduced surface expression of complement regulatory proteins CD55 and CD59 has been found in leucopenic patients with SLE [11, 12]. Deficiency of these proteins may make these cells susceptible to complement-mediated lysis. There is increasing evidence that endogenous production of type 1 interferons (IFNs) is implicated in the pathogenesis of neutropenia and lymphopenia in SLE. Elevated serum levels of IFN-a in SLE correlate inversely with leucocyte numbers [13,14].
Immunosuppressive agents like azathioprine or cyclophosphamide have the potential to worsen leukopenia via bone marrow suppression, which is less common.
Neutropenia
It is a common feature of SLE, with a prevalence rate of 47%, it may be mediated by anti-neutrophil antibodies. Increased levels of TNF-related apoptosis-inducing ligand (TRAIL) in SLE may contribute to neutropenia through excessive neutrophil apoptosis mediated neutropenia [15].
Other causes for neutropenia development in SLE are drug induced such as immunosuppressant, concomittent edications other than corticosteroids, immunosuppressives or NSAIDs [16].
Decreased Eosinophils and Basophils
Steroid therapy may cause low absolute eosinophil and monocyte counts. The basophil counts may also be decreased in SLE, particularly during active disease [9].
Thrombocytopenia
It has a reported prevalence ranging from 7 to 30% in large series of patients with SLE [14]. Increased peripheral destruction of platelets and presence of anti-platelet antibodies, is the most likely pathogenic mechanism. Presence of antiphopholipid autoantibodies in some patients has a role. Antibodies against thrombopoietin, the thrombopoietin receptor c-Mpl and CD40L have been found in some thrombocytopenic patients with SLE [14].
Thrombocytopenia in SLE may be acute in onset and extremely severe which is usually related to active disease and responds to corticosteroids. The chronic form is more common and is less likely related to disease activity; it is typically less responsive to steroid therapy.
Immune thrombocytopenia (ITP) may predate SLE in up to 16% of patients, appearing up to 10 years before SLE becomes clinically apparent [14,17]. It has been estimated that 3-15% of patients with ITP develop SLE. Low baseline levels of complement C3 were found in some thrombocytopenic patients with SLE. This correlates with an increased risk of relapse in thrombocytopenia in these patients and predicts subsequent C3 measurements. Patients with a history of thrombocytopenia and a low complement level are likely to experience relapse in thrombocytopenia, regardless of other system flares.
Thrombocytopenia is an independent risk factor for increased mortality in SLE. In a retrospective study of 126 SLE patients, late-onset thrombocytopenia was associated with an increased mortality [2]. In a more recent retrospective study of 632 patients with SLE, the authors found that the prevalence of thrombocytopenia was 58% at the time of diagnosis [18]. There was an apparent association between thrombocytopenia and disease activity, increased mortality and hypocomplementemia.
TTP is a rare but life threatening complication in SLE. It was first described by Moschowitz in 1924 as a new disease characterized by the unique pathological findings of hyaline thrombi in many organs [19]. In 1966, a pentad of severe thrombocytopenia, microangiopathic hemolytic anemia, neurologic abnormalities, renal insufficiency, and fever were established [19]. These symptoms may, also present in SLE relapses. It is important to distinguish between the two diseases because of the therapeutic implications. Recently, a mechanism for platelet consumption in TTP has been elucidated. A deficiency of ADAMTS-13 (a disintegrin-like and metalloprotease with thrombospondin type 1 repeats, a specific von Willebrand factor-cleaving protease) or an autoantibody directed against it is responsible for some cases of TTP, leading to platelet aggregation and thrombosis. Detection of the fragmented peripheral red blood cells helps in early diagnosis of TTP. Severe ADAMTS13 deficiency and the presence of an inhibitor to ADAMTS13 may be highly specific for TTP and it indicates poor prognosis, delayed response to plasmapheresis, higher plasma volume requirement to achieve complete remission and more frequent flare-ups. High titers of anti-ADAMTS13 antibodies may be associated with a more advanced stage or refractory disease [19].
Moreover, thrombocytopenia in SLE can be a complication of immunosuppressant therapy such as azathioprine and it is rarely caused by antimalarials such as hydroxychloroquine.
Pancytopenia
It may results from bone marrow failure, such as in the case of aplastic anemia. Macrophage activation syndrome, although unusual has been reported in SLE. It is manifested by fever, weight loss, arthritis, pericarditis, rash, myocarditis, nephritis, splenomegaly, hepatomegaly, lymphadenopathy, anemia, leukopenia, hyperferritinemia, anti-DNA antibodies, low CRP (< 30 mg/L) and hypocomplementemia [9].
Leukocytosis
Mostly granulocytosis, is usually due to infection or the use of high doses of steroids, but may occur during acute exacerbations of SLE. A shift to more immature forms (a "left" shift) suggests infection [9].
Lymphadenopathy and Splenomegaly
Enlargement of lymph nodes occurs in approximately 50% of patients with SLE. It is more frequently noted at disease onset or during exacerbations. Lymphadenopathy can also be due to infection or a lymphoproliferative disease in SLE such as angioimmunoblastic T cell lymphoma which is characterized by arthritis, Coombs' positive hemolytic anemia, skin rash, fever, and weight loss [9].
Splenomegaly occurs in 10%-46% of patients, particularly during active disease. Pathologic examination of the spleen reveals an onion skin appearance of the splenic arteries, a lesion that is thought to represent healed vasculitis. The possibility of lymphoproliferative malignancy needs to be considered well in SLE patients with lymphadenopathy and/or splenomegaly as the risk of non-Hodgkin lymphoma is increased four to five fold in SLE [9].
AN APPROACH TO THE INVESTIGATIONS OF HEMATOLOGICAL DISORDERS IN SLE
A detailed history is essential, including symptoms of anemia, bleeding tendencies, and particular attention should be given to drugs such as statin, antibiotic and angiotensin converting enzyme inhibitor use. Leucopenia and thrombocytopenia may complicate treatment with azathioprine, methotrexate and rarely, cyclosporine mycophenolatemofetil or hydroxychloroquine. Neutropenia may follow pulsed cyclophosphamide. Macrophage activation syndrome should be considered if cytopenia develops rapidly, especially in juvenile SLE. If neutropenia with pyrexia >38.0º C is present, blood cultures and samples from other sites should be sent for microbiological examination. In cases of pancytopenia or suspected pure red cell aplasia, parvovirus B19 serology should be performed [9.14]. Reticulocyte index is the major step for determination of the cause of anemia [9]. If the reticulocyte count is high, a hemolytic process or acute bleeding should be suspected, if it is low, anemia of chronic disease or nutritional deficiencies namely IDA, folate and vitamin B12 deficiency should be suspected (Fig. 2). Examination of peripheral blood film stained with Wright's stain provides clues in the diagnosis of anemias, leucopenia and thrombocytopenia.
Other tests are specific according to the presenting problems. Ferritin is necessary for diagnosing IDA. Concentrations greater than 20μg/dL, excludes IDA and a bone marrow examination may be considered; however, because ferritin is an acute phase reactant, it can be elevated in any inflammatory process of any cause.
Direct Coomb’s test, serum lactate dehydrogenase, liver function tests, immunoglobulins and serum protein electrophoresis have to be measured in AIHA [9].
Peripheral blood gene rearrangement studies (immunoglobulin heavy chain gene or T-cell receptor gene rearrangement) should be considered if there is a high index of suspicion of a lymphoproliferative disorder.
Measurement of anti-platelet antibodies, mainly directed against glycoproteins IIb/IIIa and Ib/IX are helpful in cases of severe and refractory thrombocytopenia [14].
IgG and IgM anti-neutrophil and anti-lymphocyte antibodies may also be measured using flow cytometry to determine the cellular specificity and antibody immunoglobulin class.
Bone marrow examination should be considered in all cases of severe or persistent leucopenia or thrombocytopenia in SLE and in pancytopenia, particularly if the patient is receiving myelotoxic therapy such as azathioprine, mycophenolate or cyclophosphamide. Specific features may present in the marrow to suggest drug-induced myelotoxicity [14].
MANAGEMENT
Anemia
Asymptomatic ACD does not require specific treatment, but if symptoms develop due to ACD and there is no indication for steroids or other immunosuppressive therapy, an erythropoiesis-promoting agents should be given such as epoetin alfa (recombinant human erythropoietin) or darbepoetin alfa, a unique molecule that stimulates erythropoiesis with a longer half life than recombinant human erythropoietin. If patients do not respond to erythropoiesis promoting agents consider administering steroids at high doses (prednisolone 1mg/kg per day in divided doses, or its equivalent). If the response is unsatisfactory (hemoglobin still <11g/dL) approximately one month after initiating treatment, the dose should be reduced and discontinued if there is no other indication for its use, but if response is good, steroid dose should be tapered to the lowest dose that maintains the improvement. Immunosuppressive also may help, but they carry a risk of further bone marrow suppression.
In the absence of active inflammation, erythropoiesis-stimulating agents may be indicated in patients with anemia due to renal insufficiency if they present with symptomatic anemia or hemoglobin concentration <11g/dL.
PRCA responds to steroids, although cyclophosphamide and cyclosporine have been successfully employed. In aplastic anemia, immunosuppressive therapy may be effective.
AIHA responds to steroids (1mg/kg per day prednisone or its equivalent in divided doses) in 75 to 96% of patients. Once the hematocrit begins to rise and the reticulocyte count falls, steroids can be rapidly tapered. If there is no response, consider pulse steroids (1000 mg methylprednisolone intravenously daily for three days), azathioprine (up to 2 mg/kg per day), cyclophosphamide (up to 2 mg/kg) or splenectomy which has success rates as high as 60% in some cases. Intravenous immunoglobulin, danazol, mycophenolate and rituximab are other options for refractory AIHA [9].
Thrombocytopenia
Treatment should be considered if bleeding or severe bruising are present, or platelet counts <50x10⁹/L. In cases of severe thrombocytopenia (<20x10⁹/L), treatment with prednisolone in tapering doses (from 1mg/kg/day) should be commenced. Pulsed methylprednisolone may also be used. It was thought that there is probably no advantage in the use of pulsed methylprednisolone over high-dose oral steroids, however; the risk of serious adverse effects such as avascular necrosis is increased [21,22]. Treatment with four to eight cycles of oral high dose dexamethasone (40 mg per day for four days) at intervals of two to four weeks may result in similar remission rates and better long-term responses than those treated with daily prednisone. Presence of antibodies against the TPO receptor may be associated with a poorer response to steroids. Most patients respond to steroid therapy within one to eight weeks. If there is no significant increase in the platelet count within one to three weeks or side effects are intolerable, other treatments should be considered [9,14].
Azathioprine is a steroid-sparing agent used in treating SLE thrombocytopenia [23,24]. Cyclosporin is an alternative immunosuppressive drug to treat SLE cytopenias [25]. In an open-label trial on 16 lupus patients, platelet and leucocyte levels returned to normal in those who were treated with cyclosporin 3-5 mg/kg/day for an average treatment period of 30 months [26], but as the use of cyclosporine carries the risk of nephrotoxicity, the preferred doses are up to 2.5 mg/kg/day, lower doses with close monitoring of renal function and blood pressure are beneficial. Its success as a steroid-sparing agent in lupus associated thrombocytopenia, has been reported [27].
Treatment with a combination of prednisolone and hydroxychloroquine may be an adequate alternative for controlling thrombocytopenia in many patients [28].
If treatment with prednisolone or steroid-sparing agents is unsuccessful, splenectomy should be considered which has been shown to produce good response [29,30]. Appropriate prophylactic measures against infection should be considered with the use of pneumococcal, haemophilus influenzae type B and meningitis vaccines and prophylactic antibiotics such as penicillin V following splenectomy, particularly in patients with additional chronic hypocomplementemia, as there might be risk of a flare in disease activity [14].
Intravenous immunoglobulins (IVIG) are well established in the treatment of ITP. They are very effective in some patients with lupus associated thrombocytopenia who show a long-lasting response to treatment [31,32]. IVIG down regulate autoantibody production, neutralize pathogenic autoantibodies by anti-idiotypic antibodies, inhibit complement-mediated damage, modulate cytokine production, induce apoptosis in lymphocytes and monocytes and modulate both B and T lymphocyte function. The therapeutic dose in severe SLE thrombocytopenia is 2g/kg, for five consecutive days.
Danazol is generally safe and well tolerated and like IVIG can be used in pregnancy [33,34].
Refractory Cytopenias and TTP
Severe and refractory cytopenias and aplastic anemia in SLE may necessitate treatment with more potent cytotoxic drugs. Oral cyclophophamide in doses of 0.75-1.0 g/m² body surface area or intravenous 10-5 mg/kg, intravenously monthly for at least 4 months appears useful in severe lupus-associated thrombocytopenia refractory to standard therapies [35]. Lower doses given more frequently, for example every 2 weeks, are used to improve tolerability without loss of efficacy. High dose intravenous cyclophosphamide has been successfully used in treating aplastic anemia complicating SLE, concurrent use of recombinant human G-CSF and antibiotics are needed. Plasmapheresis has been used to treat refractory autoimmune thrombocytopenia, PRCA, hemophagocytic syndrome, MAHA and pancytopenia in SLE [14, 35].
Plasmapheresis continues to be the mainstay in the treatment of patients with TTP, even when there is concomitant SLE. Mortality from TTP has decreased dramatically from 90% without treatment to 8-25% with the institution of plasmapheresis [36,20]. Other therapies that have been used include high dose steroids, cyclophosphamide and intravenous immunoglobulin. Rituximab, a monoclonal antibody against CD20 receptor, has been used in four reported cases of refractory TTP in SLE. The reported response and disease specific survival was 50% [36-38].
Mycofenolate Mofetil (MMF)
MMF, an immunosuppressive drug widely used in solid organ transplantation, is increasingly used in SLE. It is rapidly becoming an alternative therapy for remission induction and maintenance of lupus nephritis, and it is less toxic than intravenous cyclophosphomide. It is also reportedly used in the in the management of refractory hemolytic anemia and thrombocytopenia complicating SLE. It has been used in conjunction with cyclosporin in the treatment of PRCA in SLE [38]. Its use in SLE is increasing and it remains a promising therapy for the management of its hematological manifestations [39,40].
Anti-B-Cell Therapies
Rituximab is a chimeric monoclonal antibody against human CD20. It rapidly depletes peripheral blood CD20 positive B cells via complement-mediated and antibody-dependent cellular cytotoxicity. In SLE patients, rituximab has been shown to deplete autoreactive B cells and reduce autoantibody production by plasma cells [41-42]. Its use in severe thrombocytopenia showed a favourable response in stabilizing platelet numbers over 100x10³/l for > 6 months and disappearance of anti-DNA antibodies. Rituximab has also been used successfully in treating AIHA in SLE [14]. Overall, current data suggest that rituximab is a promising option for the treatment of severe hematological manifestations in SLE. Belimumab, is a fully human monoclonal antibody that binds to autoreactive B-cell survival factor B-lymphocyte stimulator (BLyS) with high affinity. It neutralizes human BLyS bioactivity in vitro and in vivo and it has been tested in a Phase I trial that included 70 SLE patients and was found to be safe with no clinically significant differences from placebo in adverse events. It also significantly reduces circulating B cells and anti-dsDNA antibodies. Phase II trial includes 449 patients with SLE, it was found that there was a longer time to disease flare in patients receiving belimumab compared with placebo, while a reduction in anti-dsDNA titres was also noted. In a subgroup analysis, patients with serologically active disease responded significantly better to belimumab therapy [43].
Other Approaches
Eltrombopag is a thrombopoeitin (TPO) receptor agonist that activates TPO surface receptor on the megakaryocytes which results in increased platelets production. It is approved for treatment of ITP. This drug is being used successfully in some patients with SLE who had refractory ITP and appear to be effective as a rapidly acting corticosteroid sparing therapy for patients with ITP associated with SLE [44]. One retrospective review of 3patients with SLE-associated ITP refractory to treatment with corticosteroids and other immunosuppressive therapy, were treated with eltrombopag at adose of 50 mg daily, they maintain acceptable platelet counts >50,000/mL for >6 months following tapering and cessation of corticosteroids dosage. The drug was well-tolerated and there were no adverse events [44].
Romiplostim (AMG 531) is another TPO receptor agonist that is licensed for the treatment of chronic refractory ITP. It has no sequence homology with TPO, hence there is less risk of developing antibodies against endogenous TPO. It is administered as weekly subcutaneous injections and the response is dose-dependent, peaking at Days 12-15 [14,45]. Litrature review revealed a case report of successful treatment of a pregnant woman with SLE at 27 week of gastation, she developed severe thrombocytopenia presented with bleeding at multiple sites. Her thrombocytopenia was resistant to most treatment modalities, including steroids, IVIG, immunosuppressives and even Rituximab. The addition of Romiplostin to her results in an adequate response of platelets count and control of bleeding with improvement of thrombocytopenia [46].
Antiphospholipid Syndrome (APS)
APS was first described in 1980 in patients with SLE who had increased titers of aCL with the clinical features of recurrent thrombosis, thus defined as secondary APS. But later on, by 1985 it had become apparent that some of these patients exhibited no features of underlying connective tissue disease and the concept emerged that APS could exist as a primary syndrome [47]. Systemic clinical manifestations such as central nervous system, renal and skin involvement, in addition to laboratory markers such as antinuclear antibodies and persistent thrombocytopenia may define a subgroup of primary APS closer to full-blown SLE. It was noted that we may deal with:
Patients initially classified as Primary APS gradually developing SLE, or, - Patients with SLE and associated APS, or, - Patients with SLE and positive antiphospholipid antibodies without APS manifestations [48].
In a cohort of 1000 patients with APS, 53.1% had primary APS (P-APS), while 36.2% had APS associated with SLE (SLE-APS) and with other diseases in 5.9% [49].
Revised Sapporo Classification Criteria for APS
APS is present if at least one of the following clinical criteria and one of the laboratory criteria are met: Clinical criteria: Presence of either vascular thrombosis or pregnancy morbidity, defined as following: 1- Vascular thrombosis: One or more episodes of venous, arterial, or small vessel thrombosis in any tissue or organ. Thrombosis must be confirmed by objective validated criteria, i.e. unequivocal finding of appropriate imaging studies or histologic evidence of thrombosis without inflammation in the vessel wall. 2- Pregnancy morbidity: - One or more unexplained death at or beynod the 10th week of gestation of a morphologically normal fetus, with normal fetal morphology documented by ultrasonography or by direct examination of the fetus, OR - One or more premature births before the 34th weeks of gestation of a morphologically normall neonate because of eclampsia or severe preeclampsia, or placental insufficiency, OR - Three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, that are not explained by maternal or paternal chromosomal abnormalities or by maternal anatomic or hormonal causes. Laboratory criteria: The presence of aPL, on two or more occasions at least 12 weeks apart and no more than five years prior to clinical manifestations, as demonstrated by one or more of the following: - Lupus anticoagulant (LA) activity in plasma, detected according to the guidelines of the ISTH (Scientific Subcommittee on Las/phospholipid-dependent antibodies). - aCL antibody of IgG and/or IgM isotype in serum or plasma, in moderate or high titer (>40 units GPL or MPL or >99th percentile), measured by a standardized ELISA. - Antibodies to ß2-glycoprotein I (anti-β2GPI) of IgG and/or IgM isotype in serum or plasma (at a titer >99th percentile), measured by a standardized ELISA [50,51].
Catastrophic antiphospholopid syndrome (CAPS), is rare and comprises less than 1% of APS cases and is associated with multiple, widespread vascular occlusions, high titer aPL antibody and significant mortality. Patients may develop multiple vascular occlusions in a short period of time. CAPS is frequently fatal, with a reported mortality rate approaching 50% despite anticoagulant and immunosuppressive treatment [52].
CAPS may affect SLE-APS patients with female to male ratio of 8.5:1 as compared to 1.5:1 in P-APS, it occurs at significantly younger age. In a cohort of 103 patients with SLE who had CAPS (SLE-CAPS) from CAPS registry, it was found that CAPS was the first manifestation of APS in 46% SLE patients, there were precipitating factors in 61% of patients, among them infections were present in 29%, surgical procedures in 10%, active lupus (flare) was present in 9%, obstetrical conditions in 6%, while in 39% of cases, the precipitating factors were unknown. Cerebral involvement was more likely in SLE-CAPS. This cohort showed that the administration of cyclophosphamide to patients with SLE-CAPS was associated with increased survival, whereas it had a worsening effect on the prognosis of patients with P-CAPS. The presence of lupus in patients with CAPS is a poor prognostic factor as regard to the mortality in CAPS which can be attributed to increase lupus activity and organ damage that already been there at the time of CAPS event [52].
Management of APS
Treatment of different manifestations of SLE-APS or secondary APS is the same as that of P-APS. However, other issues that need to be considered are:
Hydroxychloroquine (HCQ)
HCQ addition in the treatment of patients with SLE, APS either primary or secondary is associated with decrease aPL titers and decrease risk of thrombosis [53]. HCQ inhibits the aPL-induced expression of platelet GPIIb/IIIa receptor (platelet activation) in a dose-dependent fashion [54]. One study showed that HCQ reverse the binding of IgG-B2GPI complexes to phospholipid bilayers and cells, thus reducing thrombotic events in APS patients [55].
Rituximab
Treatment with rituximab results in B cell depletion. A litrature review of 21 patients with APS, showed the resolution of APS clinical manifestation in 19 patients, 2 responded but had recurrence of manifestations later (1had deep vein thrombosis and the other had thrombocytopenia), 2 did not respond to rituximab, the serological outcomes were reported in 12 patients, and in 10 of them, aPL either became negative or decreased significantly [56]. A systematic review of the off-label use of rituximab in APS revealed the high rate of therapeutic response in patients with APS (92%) [57,58]. One case report of CAPS with pulmonary manifestation had showed the effectiveness of rituximab in inducing remission and improvement of other life threatening complications in catastrophic APS [59].
Autologous Hematopoietic Stem Cell Transplantation (HSCT)
Autologous HSCT appears to be a promising therpy in SLE-APS, as shown in one study conducted on 46 patients with severe and refractory SLE and APS, prevalence of APS was 61%. 10 patients had positive LA, and 14 patients had high titers of aCL antibodies immediately before the stem cell transplantation. LA disappeared in 75% of patients after HSCT, aCL IgG titers were normalized in 71% of patients, while aCL IgM became and remained negative in 82% of patients. 82% of patients with SLE-APS discontinued anticoagulation a median of 4 months after the transplantation, 78% remained thrombosis free for up to 78 months (median 15 months) after HSCT. All 46 patients with SLE treated by HSCT, had shown decreased levels of ANA titers [60].
Moreover, treatment SLE-APS patients with HSCT and rituximab appears to be promising also, but further investigation and more clinical trials are required to evaluate the safety profile of these drugs, to identify the patients suitable for these aggressive therapeutic approaches and to know the effect of these approaches as long term cures of APS. Refractory APS, whether primary or secondary and catastrophic APS could be the appropriate targets for these therapies currently [61].
CONCLUSIONS
- Hematological abnormalities are the commonest among all other manifestations in SLE, and their treatment is challenging.
- Thrombocytopenia is an independent risk factor for increased mortality in SLE.
- Bone marrow examination should be considered in all cases of severe or persistent leukopenia or thrombocytopenia in SLE, to exclude dug-induced myelotoxicity in susceptible patients.
- TTP can mimic SLE relapse, therefor detection of fragmented RBCs on peripheral blood smear is mandatory for its diagnosis and treatment with plasmapheresis.
- Refractory cytopenias and aplastic anemia in SLE necessitate treatment with potent immunosuppressive drugs.
- Anti B cell therapies with Rituximab and Belimumab are encouraged and increasingly used in severe and refractory cases of different hematological disorders in SLE. Their use is associated with moderate reduction in anti-ds DNA titers, therefore significant improvement and encouraging results. * Mycophenolate Mofetil, a less toxic drug than CYC, is successfully used for the management of refractory hemolytic anemia and thrombocytopenia in SLE, therefore, it remains a promising therapy for the management of hematological manifestations in SLE patients.
- Furthermore, a number of novel approaches for cytopenia management in SLE including thrombopoeitin receptor agonists, show great promise.
- Treatment of SLE-APS and Catastrophic APS with rituximab and HSCT is promising, but more clinical trials are needed to evaluate the effects and safety profile of these therapies in the treatment of targeted patients.
CONFLICT OF INTEREST
The author declares that they have no conflict of interest with respect to the authorship and/or publication of this article.
ACKNOWLEDGEMENTS
The work to produce this review article is supported by Alzaidi's Chair of research in rheumatic diseases, Umm AlQura University, Makkah.

