All published articles of this journal are available on ScienceDirect.

New Insights into the Mechanism of Notch Signalling in Fibrosis
Authors Info & Affiliations
Abstract
The Notch pathway is an evolutionary conserved signalling mechanism that regulates cellular fate and development in various types of cells. The full spectrum of Notch effects has been well studied over the last decade in the fields of development and embryogenesis. But only recently several studies emphasized the involvement of the Notch signalling pathway in fibrosis. This review summarizes the structure and activation of the Notch family members, and focuses on recent findings regarding the role of Notch in organ fibrogenesis, in humans and in animal models.
INTRODUCTION
The treatment of tissue fibrosis is a challenge for the medical community. Indeed, 40 % of the developed world mortality is caused by fibrotic diseases and, to date, no efficient therapy is available. Over the past two years, an increasing number of papers have reported the involvement of new mediators in fibrogenesis, including the Notch-signalling pathway. In the present review, we will first present the Notch family members, their functions and structures. Then, we will focus on the implication of Notch-signalling in fibrosis and develop its role in the development of fibrotic diseases affecting various organs especially in the context of rheumatic diseases.
STRUCTURE, ACTIVATION AND FUNCTIONS OF NOTCH RECEPTORS AND LIGANDS
Notch proteins are single-pass transmembrane receptors with a conserved expression among animal species during evolution. Their principal function is the regulation of many developmental processes, including proliferation, differentiation and apoptosis [1]. Four Notch proteins have been described in mammals (Notch 1 to 4), and they have non-redundant functions during embryogenesis [2].
Structure of Notch Receptors and Ligands
Notch receptors are located at the cell surface with extracellular and intracellular portions linked in a non-covalent manner (Fig. 1). The binding of the extracellular portion to its ligand triggers two successive proteolytic cleavages in the receptor. The second cleavage leads to the release of the intracellular portion of the receptor in the cytoplasm and its translocation to the nucleus. The Notch extracellular domain is characterized by numerous EGF-like repeat domains (29 to 36) that are critical for the binding of the ligand [3]. EGF-like repeats are followed by three cystein-rich domains (LIN) that prevent signalling in the absence of ligation [4]. The intracellular portion of Notch receptors (NICD, Notch Intra-Cellular Domain) contains regions which mediate signal transduction: a RBP-J-associated-molecule (RAM) domain, ankyrin repeats (ANK) which interact with downstream proteins, a transactivation domain (TAD) and a C-terminal PEST (Prolin, Glutamic acid, Serin, Threonin) domain that is pivotal for the stability of the protein. Vertebrates and mammals have four different Notch receptors, that differ mainly in the number of EGF-like repeats and C-terminal sequences located between the ANK and PEST domains.
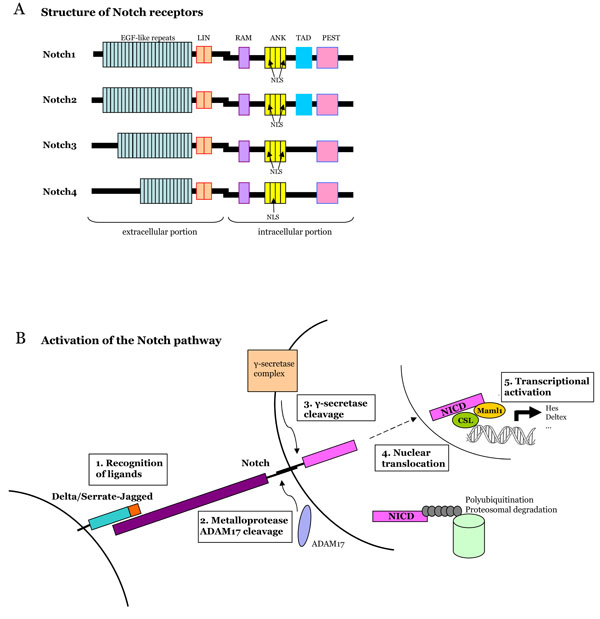
A. Structure of Notch receptors in mammals. Notch is a heterodmeric transmembrane receptor. The extracellular domain containsEGFlike repeats and LIN repeats. The intracellular domain contains a RAM domain (RBP-J associated molecule), nuclear localization sequences (NLS), seven ankyrin repeats (ANK), a transactivation domain (TAD) and a PEST sequence (Proline, Glutamate, Serine, Threonine-rich domain). The four Notch receptors differ in the number of EGF-like repeats and Cterminal sequences. B. Activation of the Notch pathway. Notch binds its l igand, is first cleaved by a metalloprotease and second by the γ-secretase compex. These cleavages allow the release of the Notch intra-cellular domain (NICD) and its transport into the nucleus, where it cooperates with transcription factors to regulate gene activity.

Hypothetical modes of activation of the Notch pathway in systemic sclerosis. ROS produced during the ischemiareperfusion phenomena or through the injection of HOCl in mice, could activate the metalloprotease ADAM17, thus indirectly triggering the release of NICD. NICD can then translocate to the nucleus and activate the transcription of its target genes leading to the development of fibrosis.
Notch ligands are transmembrane proteins with a large extracellular portion. They are encoded by genes of the Jagged (JAG1 and JAG2) and Delta-like (DLL1, 3 and 4) families [4]. Each ligand contains EGF-like domains and a “Delta-Serrate-Lag” (DSL) sequence, conserved among Drosophila melanogaster, Caenorhabditis elegans and vertebrates (Fig. 1). Depending on the context, the ligand can be produced by a cell from the same lineage as the cell expressing Notch receptor or from a disctinct population [5]. Also, ligands can interact with Notch within the same cell (cis-interactions), and inhibit Notch signalling [6]. The mechanisms leading to the expression of an active ligand on the cell surface are still not clear, and further molecular studies are needed.
Activation of Notch Receptors
The activation of the Notch-signalling pathway requires cell to cell contact. The binding of Notch to one of its ligands triggers an extracellular cleavage by the ADAM17 metalloprotease (also named TACE, for Tumor-necrosis-factor Alpha Converting Enzyme), thus leading to the formation of a membrane-tethered cleaved form of Notch (called NEXT), which is not the active form of the receptor [7]. This first step is followed by a second cleavage achieved by a γ-secretase complex [8]. The Notch cleavage by the γ-secretase enzymatic complex allows the release of the intracellular domain of Notch (NICD), the active form of Notch proteins [9]. The γ-secretase complex is the target of DAPT, a pharmacological inhibitor of the Notch pathway used in several preclinical studies mentioned below. NICD translocates to the nucleus where it induces target genes, that belong mainly to the Hes and Hey family [1, 2].
The release of the intracellular portion of Notch receptors is a key-step in the activation of the Notch pathway because NICD is directly involved in the transcriptional regulation of nuclear target genes, without relying on second messengers or phosphorylation cascades [5]. Not only is the activity of Notch receptors regulated by ligand binding and proteolytic cleavages, it is also regulated through post-transcriptional mechanisms such as glycosylation, endocytosis, endosomal trafficking and recycling, and ubiquitination [3, 5].
Functions of Notch Receptors
Notch-signalling controls the developmental fate and differentiation of cells. Thus, mutations of Notch receptors and ligands in mice lead to dysfunctions in many tissues, including the vascular system and the immune system. In humans, mutations of genes coding for different members of the Notch pathway have been linked to hereditary diseases, including the Alagille syndrome, CADASIL syndrome (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leucoencephalopathy), tetralogy of Fallot, and spondylocostal dysostosis [10, 11]. Moreover, deregulation of Notch signalling can lead to cancer [12]. The involvement of Notch in the development of haematological malignancies, such as T-acute lymphoid leukemia, is now established, and recent studies have reported a role for Notch in solid tumours [11]. Indeed, depending on the cell type and context, Notch can act as a tumor promoter or suppressor [12].
NOTCH AND EPITHELIAL-MESENCHYMAL TRANSITION (EMT)
Epithelial-mesenchymal transition is a process, initially described in early embryogenesis, through which epithelial cells acquire a mesenchymal-like phenotype [13]. Epithelial cells lose their epithelial characteristics, including E-cadherin expression and apical-basal polarity, and reorganize their cytoskeleton to acquire a motile behaviour and the phenotype of myofibroblasts. This dynamic process has been well studied in the embryonic development, but also plays a key role in the genesis of new fibroblasts during the development of organ fibrosis in adult tissues. Indeed, in mature tissues, epithelium can undergo EMT following epithelial stress such as inflammation or wounding, leading to fibroblast proliferation and fibrogenesis [14]. Thus, epithelia contribute to fibrosis by creating new fibroblasts that coexist with resident fibroblasts to produce extracellular matrix in excess.
A large bulk of evidence suggests that EMT is associated with fibrotic diseases such as progressive chronic kidney disease, lung fibrosis and, possibly, liver fibrosis [14, 15]. Recently, several studies have shown that the Notch pathway is involved in the EMT induction process [16, 17]. In 9.5 days old mouse embryos Timmerman and colleagues demonstrated that Notch is critical for the promotion of EMT in the developing heart. In this study, mice with a targeted deletion of Notch1 receptor or its effector RBP-JK-CSL exhibited abnormal maintenance of intracellular endocardial adhesion complexes and abortive endocardial EMT in vivo and in vitro. In another recent study [17], Aoyagi-Ikeda and colleagues have shown in RLE-6TN cells (rat alveolar epithelial cells) that the activation of the Notch pathway by ectopic expression of NICD or by co-culture of RLE-6TN cells with Jagged-1, induces the expression of α-SMA, a marker of myofibroblasts, type-1 collagen and vimentin. They showed that the Jagged1/Notch signalling pathway induces epithelial-to-mesenchymal transition by interactions with the TGF-β pathway. Indeed, Notch induces the production of TGF-β1 and the phosphorylation of Smad3 that favours the expression of α-SMA and leads to EMT. Thus, overexpression of Notch family members can induce EMT and fibrogenesis in various organs.
NOTCH AND FIBROTIC DISEASES
Recently, several papers have suggested that overexpression of Notch signalling may have fibrogenic effects in a wide spectrum of diseases, including scleroderma [18, 19], idiopathic pulmonary fibrosis [20], kidney fibrosis [21], and cardiac fibrosis [22].
Skin Fibrosis and Systemic Sclerosis
In humans, Notch1 is expressed in all epidermal layers. Notch ligands Jagged and Delta-like are also expressed in the epidermis. Notch1 induces the differentiation of keratinocytes through the expression of early markers such as keratin1 and involucrin [23]. Notch1, 2, and 3 are also expressed in the hair follicle and are essential for its homeostasis.
Interestingly, Notch1 is also expressed in skin fibroblasts, and is able to induce the transcription and expression of α-SMA through the activation of FIZZ1 (also known as resistin-like molecule-α, RELM-α, or hypoxia-mediated inducible factor), thus triggering the differentiation of fibroblasts into myofibroblasts.
Systemic sclerosis (SSc) is a connective tissue disease characterized by vascular dysfunction, fibrosis of skin and visceral organs, and immunologic dysregulation associated with the presence of autoantibodies [24]. Although environmental and genetic factors have been incriminated, the mechanisms that are directly implicated in the pathogenesis of the disease are still unclear [25]. Some advances have been made to treat vascular complications, but no treatment has shown convincing effectiveness to reduce skin and visceral fibrosis. An increasing amount of evidence suggests that the Notch pathway is implicated in the development of the fibrosis that characterizes SSc in both rodent and human [18, 26, 27]. Indeed, Notch1 is activated in the lesional skin of SSc patients and in their fibroblasts [18, 26]. Mice with ROS-induced SSc, bleomycin-induced SSc and Tsk1-mice also display elevated levels of NICD in skin and lungs. This accumulation of NICD is associated with the overactivation of ADAM17 (TACE), a proteinase involved in Notch activation through the first cleavage of the Notch receptor [18]. Moreover, treating mice with DAPT, a γ-secretase inhibitor that blocks the release of NICD, can reduce the collagen content in skin and lungs and the production of autoantibodies, thus preventing the development of SSc in the different mouse models [18, 26, 27]. Similarly, treating SSc-mice with Notch siRNA prevented dermal thickening and fibrosis [27].
The activation of the Notch cascade has major implications on the activation of fibroblasts in SSc. Indeed, the stimulation of SSc fibroblasts with a recombinant Jag-1-Fc chimera results in their differentiation into myofibroblasts expressing high levels of a-SMA and producing high amounts of collagen and ECM [26]. The pharmacological blockade of Notch-signalling with DAPT can also normalize the proliferation rate of dermal fibroblasts extracted from lesional skin. In a wound-healing mouse model, the increase in fibroblast proliferation has been correlated with the activation of the Notch pathway and can be blocked by the treatment of mice with DAPT, showing that Notch-signalling plays important roles in the proliferative properties of fibroblasts [28].
These data emphasize the role of Notch in the fibrotic process observed in SSc in humans and in several animal models.
Several hypotheses can explain the overactivation of Notch1 in SSc. First, reactive oxygen species (ROS) could activate Notch in SSc-fibroblasts. Indeed, the involvement of ROS in the pathophysiology of SSc [29-33] has been emphasized in a large number of studies, and we have developed a model of murine SSc induced by ROS [34]. In this model, skin fibrosis is induced by intradermal generation of HOCl that oxidizes skin proteins. Among those proteins, oxidized DNA topoisomerase-1 itself can generate an oxidative stress. This stress leads to the stimulation and proliferation of fibroblasts and to the overproduction of collagen and fibrosis. It also involves endothelial cells and is, at least partly, responsible for a vasculopathy. The development of fibrosis is not limited to the skin, because DNA topoisomerase-1 and other oxidized proteins can circulate and determine a systemic fibrosis. Fig. (2) describes the hypothetical mechanisms of activation of Notch receptors in ROS-mediated SSc during this process. Intradermal ROS can also induce the synthesis of the metalloprotease ADAM17 [18, 35, 36]. ADAM17 that triggers the first step of Notch activation, that is the shedding of the ectodomain of the receptor. ROS-induced ADAM17 could be a major factor of activation of the Notch pathway in SSc. In addition, the ischemia-reperfusion process can activate HIF-1α and lead to an increase in Notch1 mRNA levels. In Ras-transformed human fibroblasts, oncogenic Ras activates Notch1-signalling, which is required to maintain the neoplastic phenotype of these cells [37]. In SSc, the hyperproliferative phenotype of fibroblasts has been associated with elevated levels of Ha-Ras and Ki-Ras [38]. Ha-Ras and Ki-Ras could activate the Notch pathway and thus trigger their differentiation into myofibroblasts with elevated proliferative capacities.
Altogether, the data obtained in skin fibroblasts both in vitro and in vivo, demonstrate a pivotal role for Notch-signalling in the development of skin fibrosis especially in patients with SSc.
Pulmonary Fibrosis
Progressive primary idiopathic or secondary pulmonary fibrosis as observed in SSc, is a rapidly progressive illness whose pathophysiology remains poorly understood. The histological lesions show fibroblast foci where mesenchymal cells proliferate and produce aberrant levels of ECM. Some studies suggest that TGF-β is involved in the recruitment of the fibroblasts [39]. Active pulmonary fibrosis is characterized by fibroblast proliferation, emergence of myofibroblasts, ECM deposition and tissue remodelling. A recent study on the abnormal differentiation of respiratory epithelial cells in idiopathic pulmonary fibrosis has reported the activation of the Notch pathway in the lung. Hes-1, a Notch target gene, was highly expressed in lung mucus cells from patients with chronic obstructive pulmonary disease, idiopathic pulmonary arterial hypertension and idiopathic pulmonary fibrosis [20].
FIZZ-1 is expressed by airway epithelial cells and alveolar epithelial cells and is endowed with fibrogenic properties [40]. In the lung, TGF-β and FIZZ-1 can induce myofibroblast differentiation and stimulate α-SMA expression in fibroblasts [41]. A study published by Liu and colleagues has shown that the Jagged1/Notch-signalling pathway is upregulated in response to FIZZ-1 and is crucial for the myofibroblast differentiation of lung fibroblasts [42]. Using FX-ko mice that exhibit deficient Notch-signalling in the absence of exogenous fucose supplementation, they showed that Notch is required for the upregulation of α-SMA induced by FIZZ-1. They used the model of bleomycin-induced pulmonary fibrosis to confirm that FIZZ-1 induces and promotes myofibroblast differentiation in vivo through Notch-signalling. Pulmonary fibrosis is associated with a TH2 cytokine response modulated in part by Jagged 1/Notch 1 signalling that regulates TH2 differenciation and the transcription of the gene of IL-4 (33). Interestingly, the induction of pro-inflammatory and pro-fibrotic factors (MCP-1, TNF-α, IL-4, TGF-β) was impaired in Notch-deficient mice.
Chronic Kidney Disease
Renal fibrosis is characterized by the increased deposition of collagen and extracellular matrix, proliferation of myofibroblasts, migration of leukocytes, dysfunctions of epithelial cells and loss of capillaries. During kidney injury, fibroblasts can originate from various sources. Recent experiments have suggested that 15 % of fibroblasts originate from bone-marrow, 35 % from local EMT involving tubular epithelial cells under inflammatory conditions, and the rest results from local proliferation [14]. Several pathways have been implicated in the development of renal fibrosis, and a growing number of data indicate that Notch-signalling plays a key role in its pathogenesis.
During kidney development, Notch1 and Notch2 are expressed but do not play redundant roles [43]. Both receptors are required for proximal tubule and podocyte developments. In mature human and rodent developed kidneys, Notch activity is not detected, indicating that the pathway is mostly silenced once kidney development is complete [44]. The reactivation of this pathway is implicated in various renal disorders in humans and in animal models. Murea and colleagues have shown that elevated levels of Notch ligands and receptors are detected in several glomerular diseases, such as membranous nephropathy, lupus nephritis, crescentic glomerulonephritis and tubulointerstitial fibrosis [44, 45]. A correlation was found between the severity of the tubulointerstitial fibrosis and the expression of cleaved Notch1 in the tubulointerstitium. Concomitantly, another study showed that Notch plays a key role in the development of tubulointerstitial fibrosis in patients and in mouse models [21]. Using pharmacologic and genetic in vivo and in vitro experiments, the authors demonstrated that the expression of Notch in renal tubular epithelial cells is necessary and sufficient for the development of tubulointerstitial fibrosis. Moreover, genetic deletion of the Notch pathway in these cells reduced renal fibrosis. A previous report had shown that transgenic mice with elevated expression of Notch1 in podocytes develop albuminuria and sclerosis of the glomerule and die by the age of 3 weeks [46].
Taken together, all these data indicate that the Notch pathway plays a key role in kidney fibrosis. Pharmacological inhibition of the Notch pathway could be an important therapeutic strategy for chronic kidney diseases and further studies are needed to determine the effectiveness of such treatments.
Cardiac Repair
Notch-signalling plays a central role in heart development. In humans, the Alagille syndrome, an autosomal dominant disorder characterized by congenital heart deficiency with pulmonary artery stenosis, enlarged right ventricle and atrial and ventricular septation defects, is linked to Jag1 and Notch2 mutations [47]. In mice, null mutants for Jag1, Notch1, Notch2 and RBPJK display multiple cardiac defects leading to early embryonic mortality [48]. In the mammalian adult heart, Notch-signalling is downregulated [49]. The mammalian adult heart responds to injury with fibrosis, and recent findings suggest that Notch-signalling is critical for cardiac repair. Increasing Notch1 signalling in mesenchymal-stem cells leads to decreased infarction size and improved cardiac function after myocardial infarction [50]. Furthermore, Russell and colleagues showed that the Notch pathway is activated in epicardial-derived cells and drives their transdifferentiation into fibroblasts after injury, through an EMT process [22]. Finally, these data provide new evidence for a role for Notch in cardiac repair.
CONCLUSION
The Notch pathway, first described for its major role in the development of organisms, is also a key mediator in fibrogenesis as demonstrated by recent studies on skin, lung, kidney and cardiac fibrosis. Further studies are needed, especially in vivo and in patients, to determine whether the inhibition of Notch ligands, receptors or downstream target genes could be effective curative strategies to treat established fibrosis.
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
Declared none.

