All published articles of this journal are available on ScienceDirect.

Regulation of Matrix Remodeling by Peroxisome Proliferator-Activated Receptor-γ: A Novel Link Between Metabolism and Fibrogenesis
Abstract
The intractable process of fibrosis underlies the pathogenesis of systemic sclerosis (SSc) and other diseases, and in aggregate contributes to 45% of deaths worldwide. Because currently there is no effective anti-fibrotic therapy, a better understanding of the pathways and cellular differentiation programs underlying fibrosis are needed. Emerging evidence points to a fundamental role of the nuclear hormone receptor peroxisome proliferator activated receptor-γ (PPAR-γ) in modulating fibrogenesis. While PPAR-γ has long been known to be important in lipid metabolism and in glucose homeostasis, its role in regulating mesenchymal cell biology and its association with pathological fibrosis had not been appreciated until recently. This article highlights recent studies revealing a consistent association of fibrosis with aberrant PPAR-γ expression and activity in various forms of human fibrosis and in rodent models, and reviews studies linking genetic manipulation of the PPAR-γ pathway in rodents and fibrosis. We survey the broad range of anti-fibrotic activities associated with PPAR-γ and the underlying mechanisms. We also summarize the emerging data linking PPAR-γ dysfunction and pulmonary arterial hypertension (PAH), which together with fibrosis is responsible for the mortality in patients in SSc. Finally, we consider current and potential future strategies for targeting PPAR-γ activity or expression as a therapy for controlling fibrosis.
INTRODUCTION
Systemic sclerosis (SSc) or scleroderma is an incurable rheumatic disease associated with early inflammation and vascular injury, followed by progressive fibrosis affecting the skin and multiple internal organs. In the diffuse form of the disease, fibrosis of vital organs contributes to substantial mortality. Fibrosis is characterized by excessive deposition of collagen and other extracellular matrix (ECM) components produced by activated fibroblasts and myofibroblasts, and has been linked to deregulated wound healing [1]. In situ transition of fibroblasts into α-smooth muscle actin (α-SMA)-positive myofibroblasts, tissue accumulation of bone marrow-derived mesenchymal progenitor cells trafficking from the circulation, and transdifferentiation of epithelial cells, vascular endothelial cells, preadipocytes and pericytes are putative mechanisms underlying the expanded pool of biosynthetically-activated mesenchymal cells that contribute to fibrosis [2]. These cellular transitions are governed by transforming growth factor-β (TGF-β), Wnts and other soluble and matrix-associated factors. This review focused on recent studies that identify the nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ) as an important novel pathway with critical roles in regulating connective tissue homeostasis. Aberrant PPAR-γ function appears to be implicated in pathological fibrosis in the skin, lung, liver, heart, kidney and pancreas. Since PPAR-γ has a fundamental role in metabolic regulation and is associated with diabetes, obesity, and lipodystrophies, these fresh insights provide a hitherto unrecognized link between metabolism and fibrogenesis.
PPAR-γ: A PLEIOTROPIC ORPHAN RECEPTOR IMPLICATED IN MATRIX REMODELING
The nuclear receptor PPAR-γ was initially identified in fat, and was implicated as the master regulator of adipogenesis [3]. Subsequent studies have delineated the pivotal role of PPAR-γ in metabolism, glucose homeostasis, inflammation and innate immunity and, most recently, regulation of connective tissue biology [4, 5]. It is now appreciated that PPAR-γ modulates connective tissue synthesis and degradation, mesenchymal cell activation, transdifferentiation and survival. Moreover, impaired tissue expression and function of PPAR-γ is associated with, and very likely contributes to, the progression of pathological fibrogenesis. Thus PPAR-γ is a potential therapeutic target in intractable fibrotic diseases such as SSc.
PPAR-γ STRUCTURE AND EXPRESSION
PPAR-γ belongs to a superfamily of nuclear receptors that serve as ligand-activated transcriptional factors regulating a wide variety of physiological processes [6]. To date, three PPAR isotypes (PPAR-α, PPAR-β/δ and PPAR-γ) have been identified that are encoded by three different genes, display differential tissue distribution, and mediate distinct biological responses involved in development, cell proliferation, differentiation and apoptosis, glucose homeostasis and lipid metabolism [7]. The human PPAR-γ gene is located on chromosome 3, and is composed of at least 11 exons (Fig. 1) [8, 9]. To date, 9 distinct transcripts utilizing four different transcription start sites have been identified, but only two functional protein isoforms known as PPAR-γ1 and PPAR-γ2 are generated [10]. Whereas PPAR-γ1 shows relatively ubiquitous tissue expression, PPAR-γ2 is largely restricted to mature adipocytes. PPAR-γ2 contains an additional 30 amino acids at the N-terminus [8].
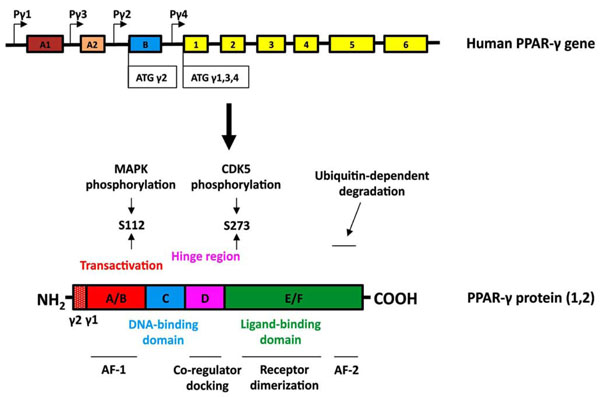
Structure of the human PPARγ gene and protein. (Upper panel, gene structure) Expression of PPAR-γ involves differential promoter usage, and the relative positions of the four known PPAR-γ promoters are designated as Pγ1-Pγ4. The transcript variants γ1, γ3, and γ4 encode the PPAR-γ1 isoform. Exon B (blue box) encodes the 28 additional amino acids found at the amino terminus of human PPAR-γ2 (30 amino acids in mouse PPAR-γ2). Exons 1–6 (yellow boxes) are common in all PPAR-γ1 transcripts and when they are spliced to exon B encode full-length PPAR-γ2. The sizes of the exon boxes approximate the relative lengths of each exon; however, the introns (depicted as straight lines) are not drawn to scale. (Lower panel, protein structure) The hypervariable A/B domain (red box) contains the activation function-1 (AF-1) domain. Human PPAR-γ2 contains a 28 amino acid amino terminal region. The C-domain (blue) contains the DNA binding domain (DBD). The D domain (hinge region, pink box) allows for conformational change following ligand binding to promote coregulator (coactivator or corepressor) docking. The E/F region (green box) contains the ligand binding domain (LBD) of PPAR-γ and the activation function-2 (AF-2) domain. PPAR-γ can be phosphorylated by MAP kinases at S112 or by CDK5 at S273. The AF2 domain participates in ubiquitin-dependent degradation and is necessary for full ligand-induced PPAR-γ transcriptional activity.
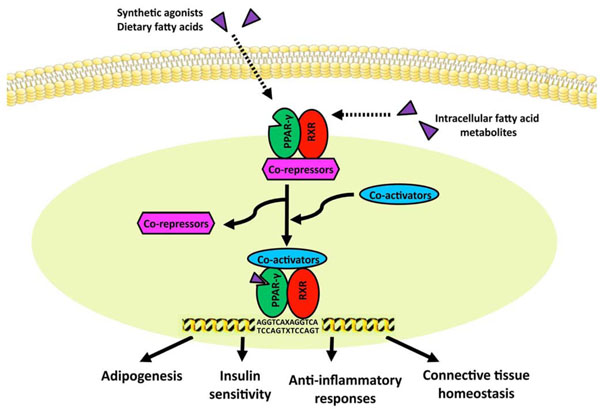
Model for ligand-induced PPAR-γ activation. In the absence of ligand, the PPAR/RXR heterodimer is bound to transcriptional co-repressors, which prevent its binding to PPRE. Upon ligand activation, PPARs undergo conformational change, and recruit co-activators such as p300/CBP and p160 to displace co-repressors, resulting in binding of target gene PPRE and inducing transcription.
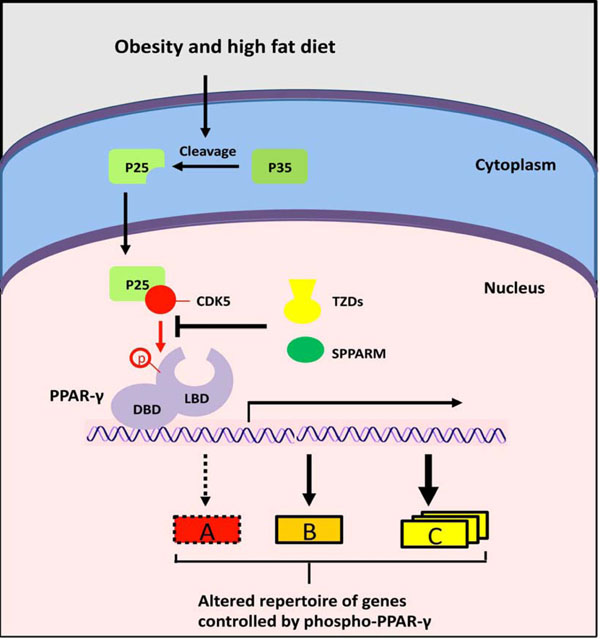
A novel mechanism for PPAR-γ signaling? Obesity and high fat diet induce inflammation and cleavage of the p35 protein to generate p25. The p25 activates CDK5, which phosphorylates PPARγ on S273, thereby preventing the transcription of specific PPAR-γ targets (A, red box). The thiazolidinediones activate the full repertoire of PPARγ target genes via binding to the LBD. SPPARMs have positive effects (green) via blocking CDK5 and stimulating a discrete number of PPAR-γ responses (A, red box) through binding to LBD.
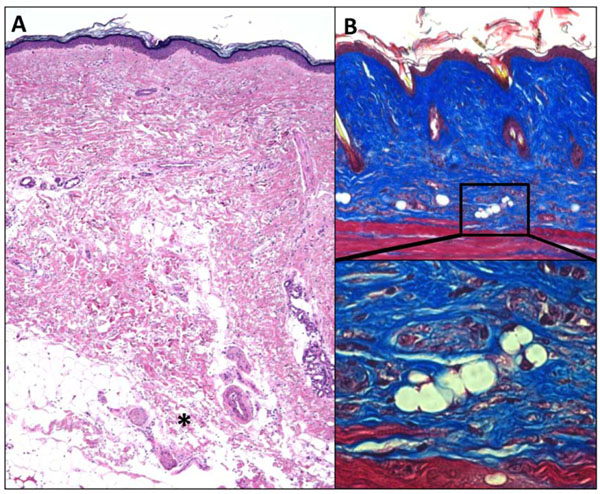
Fibrosis is associated with lipoatrophy. Skin biopsies demonstrating the association of dermal fibrosis with loss of the subjacent adipose tissue (adipose atrophy). A. SSc skin biopsy. Note dense dermal fibrosis and loss of subcutaneous fat. *, invasion of fibrotic tissue in adipose tissue. H&E stain, original magnification x 25. B. Skin biopsy from mouse with bleomycin-induced scleroderma. Note dense dermal fibrosis and absence of subcutaneous fat. Masson’s trichrome staining, original magnification x 100 (upper panel), 400 (lower panel).
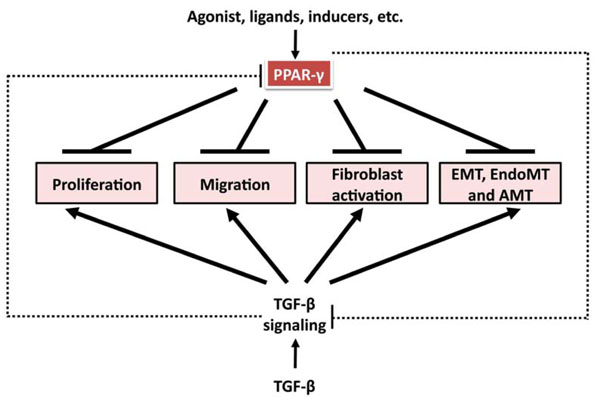
Anti-TGF-β-induced profibrotic effects of PPAR-γ ligands. TGF-β induces cell proliferation, migration, fibroblast activation, and mesenchymal transition from epithelial cells, endothelial cells, and adipocytes. All these processes contribute to fibrosis formation and progression. PPAR-γ ligands have been shown to inhibit these processes in a variety of cell types. And there is reciprocal regulation between TGF-β signaling and PPAR-γ axis. TGF-β suppresses PPAR-γ expression, which might contribute to persistent activation of TGF-β signaling; PPAR-γ ligands, through PPAR-γ-dependent and –independent mechanism, abrogate TGF-β signaling and might have therapeutic potential in fibrosis.
Regulation of PPAR-γ Expression: Transcriptional Level
In addition to abundant expression in fat, PPAR-γ is detectable in wide range of cells. Prominent among them are immune cells such as macrophages and T and B lymphocytes, as well as platelets, epithelial and endothelial cells, smooth muscle cells and fibroblasts [11]. The expression level of PPAR-γ in a given cell or tissue determines the intensity and duration of the cellular response elicited by endogenous or synthetic PPAR-γ ligands. In light of PPAR-γ's pleiotropic physiological roles, its relative abundance is predicted to have an important modulating influence on a variety of homeostatic responses. As reviewed below, a large number of soluble mediators are known to effect PPAR-γ expression, stability and activity in a tissue- and cell type-specific manner (Table 1).
Factors Regulating PPAR-γ Expression
| Negative regulators: |
|
Cytokines/growth factors/adipokines TGF-β |
| TNF-α |
| IL-1β |
| IL-13 |
| INF-γ |
| Wnt |
| CTGF Leptin |
|
others miR-27a, miR-27b Hypoxia |
| LPA |
| H3K9 methylation |
|
|
| Positive regulators: |
| C/EBPs |
| EBFs |
| ID2 |
| Nrf2 |
| Adiponectin FXR |
Natural and Synthetic PPAR-γ Ligands
| Natural agonists: |
| Unsaturated fatty acid |
| Eicosapentaenoic acids |
| Decosahexaenoic acids |
| Nitrolinoleic acids |
| Arachidonic acid |
| 15d-PGJ2 |
|
|
| Thiazolidinediones (TZDs): |
| Troglitazone |
| Ciglitazone |
| Pioglitazone |
| Rosiglitazone |
|
|
| Non-TZD dual or pan PPAR-agonists: |
| Muraglitar |
| Tesaglitazar |
| Bezafibrate |
| Tetradecyl-thioacetic acid |
|
|
| Angiotensin-II receptor blocker: |
| Telmisartan |
|
|
| SPPARMs: |
| INT131 |
| MRL24, SR1664, SR1824 |
| N-acetyl farnesyl cysteine Ajulemic acid |
Anti-Fibrotic Activity of PPAR-γ Ligands: In Vitro and In Vivo
| Ligands of PPAR-γ Inhibit ECM Gene Expression and/or Myofibroblast Transformation in Cultured Cells | Ligands of PPAR-γ Attenuate Fibrosis in Rodent Models |
|---|---|
| Hepatic stellate cells | Choline-deficient, l-amino acid-defined (CDAA) diet induced hepatic fibrosis |
| Hepatocytes | Methionine choline deficient (MCD) diet-induced fibrosing steatohepatitis |
| Hepatoma cells | Thioacetamide-induced hepatic fibrosis |
| Schistosoma japonicum induced hepatic fibrosis | |
| Renal mesagial cells | Fischer to Lewis rat renal transplantation induced chronic renal allograft damage |
| Glomerular endothelial cells | Renal injury in DOCA-salt hypertensive rats |
| Kidney fibroblasts | Diabetes associated glomerulosclerosis, tubulointerstitial fibrosis |
| BDL induced hepatic fibrosis | |
| Lung fibroblasts | Bleomycin-induced pulmonary fibrosis |
| Alveolar epithelial cells | Hypoxia induced pulmonary hypertension |
| Cardiac fibroblasts | Pressure-overload-induced cardiac remodeling |
| Cardiovascular smooth muscle cells | Cardiac fibrosis in Type 2 diabetic rat |
| Normal and Skin fibroblasts; SSc | Bleomycin induced skin fibrosis |
Cytokines and Chemokines
Several cytokines and chemokines regulate PPAR-γ expression in mesenchymal cells. TGF-β, best known as a core pathway for regulating fibroblast function, is a potent inhibitor of PPAR-γ expression in fibroblasts [12] and hepatic stellate cells (HSCs) [13]. The inhibition of PPAR-γ is mediated via the canonical Smad2/3 pathway, and two Smad-binding elements are located in the human PPAR-γ promoter [13]. Pharmacological inhibition of type I TGF-β receptor signaling and genetic disruption of Smad3 prevents the suppression of PPAR-γ by TGF-β [12]. PPAR-γ regulation is highly cell-type specific, and in contrast to fibroblasts, in monocytes and macrophages TGF-β stimulated PPAR-γ expression [12, 14]. The inflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-13 inhibit adipocyte differentiation by suppressing PPAR-γ expression [15]. In addition, interferon-γ and IL-1β are also known to suppress PPAR-γ expression [16].
Wnts
The Wnt family of secreted glycoproteins function as essential developmental signals during embryogenesis [17]. Their inhibitory effects on PPAR-γ were first recognized in 2000 when Macdougald and colleagues showed this to be the mechanistic basis for its anti-adipogenic effect [18]. Suppression of PPAR-γ by Wnt ligands involves both canonical β-catenin and non-canonical signaling pathways [18, 19]. Down-regulation of PPAR-γ is responsible for Wnt’s ability to switch adipocyte differentiation toward osteoblastic lineage [20]. Ectopic Wnt10b expression in ST2 mesenchymal progenitor cells suppresses expression of PPAR-γ as well as CCAAT/enhancer-binding protein alpha (C/EBPα), and induces osteoblast makers such as Runx2 and Dlx5 [21].
Transcription Factors
Transcription factors shown to regulate PPAR-γ expression include C/EBPs [22], EBF proteins [23], inhibitor of DNA binding (ID) 2 and NF-E2 related factor 2 (Nrf2). A recent study using high-throughput screening of inducers of adipogenesis identified harmine, a β-carboline alkaloid isolated from seeds of Peganum harmala (Syrian rue) and Banisteriopsis caapi [24]. Harmine was found to be a potent inducer of PPAR-γ expression in preadipocytes by stimulating ID2 expression [25]. Nrf2 is an inducible transcription factor that regulates genes encoding for cytoprotective and antioxidant enzymes [26]. The expression of Nrf2 is potently induced by PPAR-γ ligands in vitrovia a post-transcriptional mechanism, and Nrf2 might mediate several of the biological responses associated with PPAR-γ signaling [27]. In turn, Nrf2 binds to the PPAR-γ promoter antioxidant response element (ARE) to stimulate transcription, indicating a mutually reciprocal stimulatory relationship between PPAR-γ and Nrf2 [28].
The bile acid receptor farnesoid X receptor (FXR) is implicated in the control of hepatic glucose metabolism and peripheral insulin sensitivity. PPAR-γ ligands failed to induce adipogenesis in FXR-null fibroblasts, indicating that FXR activity is necessary for PPAR-γ expression [29]. A recent report identified the chicken ovalbumin upstream promoter-transcription factor II (COUP II) as a Wnt/β-catenin target that mediates suppression of PPAR-γ [30]. Finally, the Early Growth Response Factor-1 (EGR-1) transcription factor implicated in proliferation, differentiation and fibrogenesis [31] has a complex and cell type-specific role in regulating PPAR-γ expression. In aortic smooth muscle cells, EGR-1 mediated the stimulation of PPAR-γ expression induced by PDGF, bFGF, IL-1β [32], whereas in preadipocytes EGR-1 inhibited adipogenic differentiation by suppressing PPAR-γ [33].
Epigenetic Regulation
Epigenetic mechanisms appear to be important in regulating PPAR-γ expression via chromatin and DNA modifications, but remain incompletely characterized. Recent studies addressed the role of PPAR-γ epigenetics in hepatic stellate cells (HSCs) undergoing myofibroblast differentiation [34]. It was shown in cultured HSCs that the methyl-CpG binding protein 2 (MeCP2) is recruited to the PPAR-γ promoter, along with transcriptional repressor heterochromatin protein 1 alpha (HP1a). Consequent H3K9 methylation results in suppression of PPAR-γ expression [35, 36]. Remarkably, adipogenic loci (including PPAR-γ2 and leptin) are hypomethylated in adipocyte-derived stem cells, and hypermethylated in non-adipogenic hematopoietic cells [37]. Another study reported that induction of adipogenic differentiation in 3T3-L1 cells is associated with demethylation of the PPAR-γ2 promoter region [38]. These studies made a compelling case for the importance of DNA methylation in regulating PPAR-γ biology.
MicroRNAs (miRNA) are small non-coding RNAs that control gene expression at the post-transcriptional level by selective binding to 3’UTR. The PPAR-γ 3’UTR harbors a binding site specifically recognized by miR27a [39]. Ectopic expression of this microRNA in 3T3-L1 preadipocytes resulted in repression of adipogenic differentiation and suppression of PPAR-γ [40].
Other Modulators of PPAR-γ:
Other molecules known to inhibit PPAR-γ expression include leptin, connective tissue growth factor (CTGF or CCN2), and lysophosphatidic acid (LPA) [41-44]. Hypoxia negatively influences PPAR-γ [41, 45]. It is noteworthy that each of these soluble molecules, along with hypoxia, has been implicated in the pathogenesis of fibrosis [1]. On the other hand, adiponectin, a fat-specific adipokine whose expression is tightly regulated by PPAR-γ, itself induces PPAR-γ2 expression in a variety of cells [46].
Post-Transcriptional Regulation of PPAR-γ
PPAR-γ function is also regulated via modifications of its stability and activity (Fig. 1) [47]. Growth factors (epidermal growth factor, platelet-derived growth factor, TGF-β, insulin) stress, and prostaglandin PGF2α induce ERK1/2-mediated PPAR-γ phosphorylation on serine 112, which results in decreased transcriptional activity [48]. Overexpression of an S112 mutant PPAR-γ resulted in enhanced adipogenesis [49]. Recent studies identify serine 273 as another PPAR-γ phosphorylation site with important functional role [50]. It was shown that cyclin-dependent kinase 5 (Cdk5), which is activated in mouse adipose tissues by high-fat diet, phosphorylated PPAR-γ at S273, resulting in reduced adiponectin production. These findings stimulate re-thinking of the paradigm for the anti-diabetic mode of action of synthetic PPAR-γ ligands, which were thought to act as PPAR-γ agonists. It now appears that drugs such as rosiglitazone enhance insulin sensitivity via prevention of Cdk5-induced PPAR-γ S273 phosphorylation [50].
Ubiquitination and proteasomal degradation have important roles in PPAR-γ regulation [51, 52]. Moreover, synthetic PPAR-γ ligands themselves cause accelerated PPAR-γ degradation [51]. Therefore, agents that can block PPAR-γ degradation have therapeutic promise to enhance the efficacy of PPAR-γ agonist drugs. Indeed, a recent study using the proteasomal inhibitor bortezomib demonstrated increased tissue accumulation of PPAR-γ in mice, which may account for the anti-fibrotic activity of bortezomib in the belomycin-induced model of fibrosis (see below).
ACTIVATION OF PPAR-γ BY LIGAND
As an inducible transcription factor, PPAR-γ functions as a heterodimer with the retinoic acid-inducible retinoid X receptor (RXR). As shown in Fig. (2), the PPAR-γ/RXR heterodimer binds to PPAR response elements (PPRE), which contains repeats of the AGGTCA sequence separated by one or two nucleotides [53]. In the absence of ligand, the PPAR/RXR complex is bound to transcriptional co-repressors and histone deacetylases (HDACs), which prevent its binding to PPRE [54]. Upon ligand activation, PPARs undergo conformational change, and recruit co-activators such as p300/CBP and p160 to displace co-repressors, resulting in binding of target gene PPRE and inducing transcription.
The activity of PPAR-γ can be induced by a wide range of natural endogenous ligands and synthetic agonists (Table 2). Putative endogenous ligands include prostanoids such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and arachidonic acid derivatives synthesized through the lipoxygenase pathway such as 15-hydroxy-eicosatetraenoic acid (15-HETE) [55]. Fatty acids, eicosapentaenoic acids, docosahexaenoic acids, nitrolinoleic acid and LPA also activate PPAR-γ in a variety of cell types [56, 57]. The thiazolidinedione class of anti-diabetic drugs is the most extensively characterized PPAR-γ synthetic ligands. Their insulin-sensitizing efficacy was originally thought to involve induction of PPAR-γ transcriptional activity, resulting in up-regulation of target genes involved in lipid and glucose metabolism [58]. As noted above, exciting recent studies demonstrate that the thiazolidinedione drugs block CDK5-mediated PPAR-γ phosphorylation at S273, resulting in altered PPAR-γ function [50]. These intriguing findings open the door for the development of novel non-agonist PPAR-γ ligands that might have potent insulin-sensitizing activity while avoiding undesirable side effects associated with classic PPAR-γ agonist ligands. The glitazar family (including muraglitazar, tasaglitazar, and farglitazar) are a novel class of insulin-sensitizing drugs that activate both PPAR-γ and PPAR-α [59]. Triterpenoids are a large family of plant-derived compounds that have been used in traditional Asian medicine for centuries. CDDO, one of the most active synthetic triterpenoid derivative, shows anti-inflammatory and anti-proliferative activities. A methyl derivative (CDDO-Me, Bardoxolone methyl) showed efficacy in a Phase II clinical trial for diabetic kidney disease [60]. CDDO can bind to PPAR-γ and stimulate its activity and induce adipogenic differentiation, identifying oleanic acid-derived triterpenoids as a novel class of PPAR-γ ligands [61].
The design, synthesis and biological evaluation of novel PPAR-γ ligands with enhanced insulin-sensitizing activity but minimized side effects is the focus of intensive research [62]. Strategies for safer insulin-sensitizer drugs involve the development of selective PPAR-γ modulators (SPPARMs) that show partial or gene-selective agonist activities, and agents that can augment the expression of PPAR-γ (Fig. 3) [62]. High-throughput screening for blockers of Cdk5-mediated PPAR-γ phosphorylation led to the recent identification of SR1664, which is a potent insulin sensitizer in cultured adipocytes and in insulin-resistant mice, but completely lacks PPAR-γ agonist activity and associated side effects [63].
PPAR-γ: BEYOND ADIPOGENESIS AND INSULIN SENSITIVITY
Aberrant PPAR-γ function is implicated in an increasing number of human disorders, of which type 2 diabetes, lipodystrophy, obesity and the metabolic syndrome are best recognized [64]. Newer studies also link PPAR-γ to osteopenia, pulmonary arterial hypertension (PAH), pulmonary fibrosis, sarcoidosis, ulcerative colitis, and even aging [11, 65, 66]. Common allelic polymorphisms in the PPAR-γ gene are associated with type 2 diabetes, obesity, asthma and cardiovascular disease [67]. Dominant negative PPAR-γ mutations cause lipodystrophy and diabetes [68]. In the mouse, germline deletion of PPAR-γ results in embryonic lethality due to defective placental development [69]. Conditional deletion of PPAR-γ in the endothelium or in vascular smooth muscle cells results in spontaneous PAH [70, 71]. Moreover, treatment of mice with the PPAR-γ ligand rosiglitazone attenuates hypoxia-induced PAH [72]. Mice with conditional deletion of PPAR-γ in adipose tissue develop lipoatrophy [73]. The loss of subcutaneous adipose tissue is accompanied by marked fibroplasia in these mice resembling the subcutaneous changes observed in murine models of scleroderma and in SSc skin biopsies (Fig. 4) [74, 75].
A Novel Function of PPAR-γ: Regulation of Connective Tissue Homeostasis
Recent studies reveal an entirely novel role for PPAR-γ in tissue repair and fibrogenesis. In multiple organ-specific human fibrotic diseases, fibrosis is predictably accompanied by reduced tissue PPAR-γ levels. This has been documented in fibrosis of kidney [76], liver [77], and lung [78], and the cutaneous lesions of scarring (cicatricial) alopecia [79]. Reduced tissue expression of PPAR-γ was also found to be associated with fibrosis in various mouse models [5]. For instance, bile duct ligation (BDL)-induced liver injury and fibrosis is associated with reduced hepatic PPAR-γ expression. Genetic deletion of PPAR-γ in liver exacerbated hepatic fibrogenesis after injury [80]. Mice harboring a dominant-negative PPAR-γ mutation develop exaggerated cardiac fibrosis when challenged with angiotensin II [81]. These and similar reports suggest a causal role for reduced PPAR-γ expression or activity in the development or progression of fibrosis.
We have previously demonstrated reduced cutaneous PPAR-γ expression in mice with bleomycin-induced scleroderma [75]. Mice with fibroblast-specific PPAR-γ deletion showed increased susceptibility to bleomycin-induced scleroderma [82]. In vitro, PPAR-γ-deficient fibroblasts showed constitutive TGF-β secretion and enhanced stimulation of type I collagen and α-SMA production [82, 83]. Suppression of PPAR-γ in the skin by overexpressing Wnt10b in transgenic mice not only attenuated subcutaneous adipose tissue development, but was also accompanied by scleroderma-like skin fibrosis [84].
Impaired PPAR-γ Signaling in Scleroderma
Impaired expression of PPAR-γ has been described in SSc skin biopsies, as well as in explanted skin fibroblasts [12, 85]. The loss of cutaneous PPAR-γ in SSc may account for subcutaneous lipoatrophy that is predictably associated with the development of dermal fibrosis (Fig. 4). We and others have explored the mechanisms responsible for reduced PPAR-γ in SSc. Microarray-based expression profiling of SSc skin biopsies showed an inverse correlation between mRNA for PAI-1, a marker for TGF-β activity, and levels of PPAR-γ mRNA [12]. The most significant reduction of PPAR-γ expression was observed in SSc skin biopsies showing a robust TGF-β-activated gene expression signature [12, 86]. These same biopsies showed a reduced PPAR-γ-regulated gene signature comprising genes involved in adipogenesis and fat metabolism such as PGC1, PCK1 and FABP2, as well as tumor suppressor phosphatase and tensin homologue deleted on chromosome 10 (PTEN). These findings highlight a reciprocally antagonistic relationship between PPAR-γ signaling and TGF-ß signaling in fibrosis. Intriguingly, a trend toward a lower PPAR-γ-regulated gene signature was observed in biopsies from SSc patients with more extensive skin fibrosis, suggesting a causal role for reduced PPAR-γ expression or activity in fibrosis progression. As a marker for PPAR-γ activity, we measured serum levels of adiponectin, a direct PPAR-γ transcriptional target produced predominantly in adipocytes [87]. A significant inverse correlation between serum adiponectin levels and skin fibrosis was observed, consistent with the notion of reduced PPAR-γ activity as a risk factor for progressive skin fibrosis [88, 89]. Ethnic differences in PPAR-γ expression and function have been reported in SSc patients [90]. Whether such ethnic differences, which are likely determined by genetic variations, contribute to differing rate of disease progression or response to therapy, remain important questions to be answered.
Anti-Fibrotic Effects of PPAR-γ Ligands
Activation of cellular PPAR-γ signaling using either natural ligands or synthetic agonists, or by enhancing PPAR-γ expression, results in attenuation of profibrotic responses both in vitro and in vivo (Table 3). For instance, in cultured skin and lung fibroblasts, 15d-PGJ2 and rosiglitazone abrogated TGF-β-induced stimulation of collagen and fibronectin synthesis, myofibroblast differentiation, fibroblast migration and the secretion of profibrotic growth factors such as TGF-β and CTGF [91, 92]. Similar results have been reported in HSC [77, 93] and mesangial cells [76]. Rosiglitazone treatment of explanted SSc lesional skin fibroblasts normalized the levels of Type I collagen, CTGF and α-SMA [85]. Remarkably, PPAR-γ ligands also inhibited mesenchymal transition of alveolar epithelial cells induced by TGF-β, indicating that regulation of EMT and other forms of transdifferentiation may be a fundamental mechanism underlying the anti-fibrogenic activities of PPAR-γ ligands [94]. In addition to ligands, a constitutively active PPAR-γ construct (VP16-PPAR-γ), inhibit TGF-β-induce myofibroblast differentiation and collagen secretion in lung fibroblasts [95].
Anti-Fibrotic Effects of PPAR-γ Ligands in Animal Models of Fibrosis
Treatment with PPAR-γ ligands in a variety of mouse models, ameliorated fibrosis in the lung [95, 96], kidney [97-99], heart [100, 101] and liver [102, 103]. In bleomycin-induced scleroderma in the mouse, rosiglitazone not only prevented skin fibrosis, but also attenuated the severity of established fibrosis [75]. Hepatic overexpression of PPAR-γ delivered via adenovirus was sufficient to attenuate experimental liver fibrosis [80]. PPAR-γ activation affords protection from experimental fibrosis, as well as other forms of lung pathology. Rosiglitazone and 15d-PGJ2 attenuated pulmonary fibrosis induced by bleomycin [104]. The anti-fibrotic effects were associated with reduced inflammatory responses, making it difficult to ascertain whether the reduction of fibrosis and improved survival represented direct anti-fibrotic effects or was consequence of reduced inflammation. In a model of asthma, PPAR-γ agonist reduced lung inflammation and eosinophilia, as well as airway remodeling [105].
Loss of Endogenous PPAR-γ Antagonists is Associated with Protection from Fibrosis
Intriguing recent studies show that mice lacking endogenous inhibitors of PPAR-γ may be resistant to fibrosis. For instance, the db/db mice that have a defective leptin receptor and are resistant to leptin, which suppresses PPAR-γ, had elevated levels of PPAR-γ in lung, and were resistant to bleomycin-induced pulmonary fibrosis [106]. Fibroblasts from mPges-1 (microsomal PGE synthase-1) deficient mice showed constitutively elevated PPAR-γ levels and transcriptional activity in vitro [107], and mPGES-1-null mice were resistant to bleomycin-induced inflammation and fibrosis in skin [108]. In another promising approach, mice were treated with bortezomib, a proteasomal inhibitor, in order to block PPAR-γ degradation. Bortezomib resulted in markedly enhanced tissue accumulation of PPAR-γ presumably reflecting its impaired proteolysis. Remarkably, bortezomib-associated PPAR-γ accumulation was associated with resistance to bleomycin-induced skin and lung fibrosis [109]. Prevention or attenuation of fibrosis in rodents treated with PPAR-γ ligands or inducers not only establishes a fundamental physiological role of PPAR-γ in modulating matrix remodeling and preventing excessive fibrogenesis, but also demonstrates the feasibility of controlling fibrosis by modulating PPAR-γ expression or activity.
ANTI-FIBROTIC MECHANISMS OF PPAR-γ
The molecular mechanisms underlying the anti-fibrotic effects of PPAR-γ are the subject of intense investigation. Current studies focus on the cross-talk between the PPAR-γ axis and TGF-β signaling (Fig. 5). As master regulator of myofibroblast activation, TGF-β causes Smad2 and Smad3 phosphorylation, promoting Smad heterocomplex formation and nuclear accumulation. The Smad complex selectively binds to Smad binding element (SBE) DNA sequences, recruits coactivators such as histone acetyltransferase p300 and activates or represses transcription [110]. In addition, TGF-β also signals through Smad-independent kinase cascades including ERK1/2, PI3K/Akt, SAPK/JNK and p38 [111]. Both Smad-dependent and Smad-independent TGF-β signaling pathways have been shown to be disrupted by PPAR-γ ligands. In fibroblasts, ligand-activated PPAR-γ blocked Smad-dependent recruitment of p300 to the Col1A2 transcriptional complex [92, 112]. Because p300 promotes histone acetylation and has an essential role in the profibrotic activities of TGF-β [113], the findings imply that PPAR-γ ligands suppress fibrotic responses via epigenetic regulation. Similarly, in lung epithelial cells and lung cancer cells, PPAR-γ abrogated TGF-β-induced EMT without blocking Smad activation [94, 114]. In contrast, in hepatic stellate cells, PPAR-γ ligands blocked TGF-β-induced Smad3 phosphorylation, suggesting that the antagonistic effects of PPAR-γ ligands on TGF-β signal transduction may involve distinct cell type-specific mechanisms [115]. Fibrocytes are multipotent progenitor cells capable of differentiating into myofibroblast or into adipocyte. Treatment of fibrocytes with troglitazone disrupted TGF-β-activated SAPK/JNK signaling, leading to decreased Smad2/3 activity and myofibroblast differentiation [116]. While Egr-1, an early-immediate transcription factor important in transducing fibrogenic TGF-β activities [31, 117] regulates the expression of PPAR-γ [32], PPAR-γ ligands have been shown to block the TGF-β-induced expression of Egr-1 [118].
The anti-fibrotic activities of PPAR-γ ligands might also be indirectly mediated by its target genes such as hepatocyte growth factor (HGF) and adiponectin. In some studies, PPAR-γ stimulated the production of HGF, which is an anti-fibrotic agent that then mediates autocrine suppression of TGF-β responses [119]. Adiponectin, a fat-specific PPAR-γ target, prevents hepatic fibrosis in the mouse [120]. In hepatocytes, adiponectin induces expression of the TGF-β decoy receptor BAMBI and abrogates TGF-β signal transduction [121]. It has been therefore suggested that adiponectin may be responsible for some of the anti-fibrotic effects of PPAR-γ ligands (Feng et al., unpublished; [122]). The phosphotase PTEN is induced by PPAR-γ [123]. PTEN has potent anti-fibrotic effects and its expression is deficient in pulmonary fibrosis and in lesional SSc skin [124, 125]. Because PTEN abrogates the stimulation of collagen production and myofibroblasts differentiation [126], pharmacological stimulation of PTEN by PPAR-γ ligands may be a potential therapeutic approach in fibrosis.
PPAR-γ Ligands have PPAR-γ-Independent Anti-Fibrotic Effects
The anti-fibrotic activities elicited by PPAR-γ ligands, including inhibition of fibroblast migration, adipocyte transdifferentiation, myofibroblast transition and EMT, might also involve PPAR-γ-independent mechanism. For instance, 15d-PGJ2 and rosiglitazone potently reduced collagen production in TGF-β-treated fibroblasts was only partially reversed by a dominant-negative PPAR-γ, suggesting anti-fibrotic effects of the PPAR-γ agonists partially through PPAR-γ independent mechanisms [92]. The synthetic triterpenoid CDDO is an agonist for PPAR-γ [61]. Nevertheless, we and others have shown that CDDO inhibits TGF-β-induced myofibroblast differentiation and other fibrotic responses independent of its effects of PPAR-γ. These mechanisms include blocking PI3K-Akt signaling [127], and inducing cytoprotective Nrf2 accumulation. Finally, PPAR-γ blocks signaling via toll-like receptors (TLR), which is important in light of the role of TLRs in mediating fibrotic responses (Bhattacharyya S et al, unpublished).
PPAR-γ Ligands Negatively Regulate Wnt Signaling
The significance of Wnt signaling in fibrosis is emerging [128]. Activation of the Wnt/β-catenin pathway by Wnt ligands or by inhibiting glycogen synthase kinase 3-β in vitro or in vivo results in fibroblast activation and fibrosis [129, 130]. Thiazolidinediones induce expression of the WNT inhibitor Dickkopf-1 in adipocytes, thereby blocking profibrotic Wnt signaling and fibroblast differentiation [131]. The non-thiazolidinedione GW1929 is a PPAR-γ agonist that blocks β-catenin transcriptional activity in a PPAR-γ-dependent fashion [132]. These exciting observations indicate that blocking profibrotic Wnt/β-catenin signaling represents another mechanism accounting for the anti-fibrotic activity of PPAR-γ ligands.
REDUCED PPAR-γ EXPRESSION AND FUNCTION IN PULMONARY ARTERIAL HYPERTENSION
Pulmonary arterial hypertension (PAH) is a devastating complication of SSc, that can also occur in an isolated form, and it is associated with high mortality. A landmark study demonstrated that the expression of PPAR-γ was markedly reduced in the lungs from patients with PAH [133]. Subsequent studies have confirmed the association between impaired PPAR-γ and PAH. Moreover, targeted PPAR-γ knockout in pulmonary vessels resulted in the spontaneous development of PAH and right ventricle hypertrophy in transgenic mice [70, 71]. Pulmonary vascular remodeling was associated with marked local upregulation of platelet-derived growth factor signaling in the blood vessels. Interestingly, this process was abrogated by treatment with the tyrosine kinase inhibitor imatinib, which blocks c-Abl and PDGF receptor kinase activity. In another model of PAH induced by hypoxia, rosiglitazone attenuated vascular remodeling [134]. Therefore, PPAR-γ appears to be a fundamental molecular of pulmonary vascular homeostasis, and reduced PPAR-γ expression or function leads to PAH [135]. It is remarkable, therefore, that defective PPAR-γ expression or function appears to play a key role in the pathogenesis of both fibrosis and PAH, which two lethal complications of scleroderma, and, at least in experimental models, both of these PPAR-γ-associated complications of scleroderma could be ameliorated with PPAR-γ therapy. These findings raise hope for the future that pharmacological PPAR-γ modulation might provide an effective therapeutic strategy uniquely targeting both fibrotic and vascular complications of scleroderma.
CONCLUSION
The multifunctional nuclear hormone receptor PPAR-γ long considered a master regulator of glucose homeostasis and fat metabolism is now beginning to emerge as a regulatory molecule with fundamental roles in the pathogenesis, and possible utility in the treatment, of fibrosis in general and SSc in particular. Accumulating evidence points to PPAR-γ as the hitherto unsuspected nexus between metabolism and pathological fibrosis and vascular remodeling. Observations from acquired and genetic human diseases and animal models indicate that on the one hand, there exists a causal relationship between fibrosis (as well as pulmonary vascular disease) and defective PPAR-γ expression and function. On the other hand, these findings suggest that pharmacological targeting of PPAR-γ, by inducing its activity using ligands or CDK5 inhibitors, or by enhancing its tissue levels using proteasomal inhibitors, might be useful to prevent, and perhaps even reverse, fibrosis and vascular remodeling and PAH. Although thiazolidinedione drugs are currently used in type 2 diabetes, these PPAR-γ ligands are associated with side effects [136]. Novel PPAR-γ agonists and non-agonist ligands are in development. A better understanding of the cellular and cell type-specific mechanisms of action underlying the anti-fibrogenic and vascular remodeling activities of PPAR-γ is urgently needed. Moreover, it will be of great interest to investigate the genetic associations between PPAR-γ gene polymorphisms and specific SSc endophenotypes such as pulmonary fibrosis and PAH, as well as disease outcomes and response to therapy.
ACKNOWLEDGEMENT
We are grateful for helpful discussions with Drs Asish Ghosh, Warren Tourtellotte, Ormond A. MacDougald, Cara C. Gottardi, Anna P. Lam, Scott Budinger, Gokhan Mutlu and members of the Varga Lab. Supported by grants from the National Institute of Health (AR 42309) and from the Scleroderma Research Foundation.
CONFLICT OF INTEREST
Declared none.

