All published articles of this journal are available on ScienceDirect.

New Insights into the Role of Oxidative Stress in Scleroderma Fibrosis
Authors Info & Affiliations
Abstract
Systemic sclerosis (Scleroderma – SSc) is a connective tissue disorder of unknown aetiology characterized by extensive fibrosis of the skin and visceral organs, by vascular abnormalities and immunological manifestations.
Recent evidence suggest that the cellular redox state may play a significant role in the progression of scleroderma fibrosis. Mechanisms involved include an autoamplification circuit linking ROS, Ras and ERK 1-2 which in turn amplifies and maintains the autocrine loop made up by cytokines, growth factors and their cognate receptors.
This review summarizes the recent progress on the role of oxidative stress in the pathophysiology of scleroderma and disorders characterised by organ fibrosis
INTRODUCTION
Many chronic diseases affecting the liver, lung, skin, heart, kidney are characterized by abnormal deposition of extracellular matrix proteins and abnormal oxidative reactions. The association found between the fibrotic process and the oxidative stress has fostered the study of the mechanisms by which they influence each other. Oxidative stress may directly or indirectly stimulate the accumulation of extracellular matrix proteins. On the other hand, fibrosis may contribute to oxidative stress or both of them may be triggered by an independent mechanism. Elucidation of the signaling pathways involved in the mutual influence between these two key events is crucial for the development of adequate treatment of fibrotic disorders which are characterized by high morbidity and mortality.
Scleroderma is a severe, immune-mediated disorder the main features of which are microvascular damage, autoantibodies and fibroblast activation with fibrosis of the skin and visceral organs [1]. In spite of significant effort, the etiology and pathogenesis of scleroderma remain elusive. This gap in knowledge impairs the development of novel therapeutics able to modify the natural history of this disease.
Several years ago the pathogenesis of scleroderma was linked to the presence of a large excess of reactive oxygen species [2]. This hypothesis, over the years, has been supported by several reports which documented the presence of an abnormal redox state in scleroderma patients. The aim of this article is to update the role of oxidative stress in systemic sclerosis (scleroderma, SSc), as a paradigm of fibrotic disorders.
TERMINOLOGY
Free radicals have been defined as “any species capable of independent existence that contains one or more unpaired electron in atomic or molecular orbitals” [3]. In recent years, this definition has been substituted by the broader term of reactive oxygen species (ROS). The term ROS includes reactive oxygen intermediates (ROI) such as O2-, H2O2, OH, plus ozone (O3) and singlet oxygen (1O2) as well as hypoclorous (HOCl), hypobromous (HOBr) and hypoiodous (HOI) acids. Molecules such as hydrogen peroxide, singlet oxygen, hypoclorous acid, hypobromous (HOBr) and hypoiodous (HOI) acids are not radicals, but are capable of radical formation in the extra- and intracellular environments [4].
Excess of ROS is called oxidative stress. ROS excess may be due to abnormal generation of ROS, or to low antioxidant buffer capacity, or to decreased nitric oxide (NO) bioavailability. NO contains one unpaired electron and is therefore a radical. NO is generated in tissues by nitric oxide synthases (NOS) which generates NO through conversion of arginine to citrulline. NO reacts with oxygen and water in the extracellular space, with production of nitrate and nitrite anions. These compounds, indicated as reactive nitrogen species (RNS), must be effectively neutralized, otherwise they can trigger nitrosylation reactions that can alter the structure of proteins and inhibit their natural functions. Furthermore, nitric oxide and superoxide may react together to produce the damaging hydroxyl radical via an intermediate peroxynitrite anion which can cause DNA fragmentation and lipid oxidation.
SOURCES OF REACTIVE OXYGEN SPECIES
ROS are generated in cells by several pathways. One of the major sources is the mitochondrial electron transport chain, from which 1-2% of total electrons leak during cellular respiration. These leaked electrons contribute to the generation of O2-. Release of O2-occurs towards the cytosolic side of the inner mitochondrial membrane. However, mitocondrial O2- is kept low by mitochondrial superoxide dismutase and glutathione peroxidase, so that only H2O2, generated by these two enzymes, can permeate the mitochondrial membrane. This event is responsible for mitochondrial damage. As it happens during hypoxia, mitochondrial damage generates abnormal levels of O2- which alter ATP synthesis, mitochondrial permeability and cellular Ca++ regulation, thus predisposing the cell to death [5].
Another source of ROS is represented by the reaction of xanthine oxidase with hypoxanthine in the presence of O2, with production of superoxide, xanthine, urate and H2O2. This is an important source of ROS during ischemia-reperfusion events, when xanthine dehydrogenase is converted into xanthine oxidase by sulfhydryl oxidation [6].
Superoxide anion can also be generated by NOS. This occurs as a consequence of hypoxia, loss or oxidation of NOS cofactor tetrahydrobiopterin (BH4). The ferrous-dioxygen of the enzyme dissociates and O2- is generated from the oxygenase domain of NOS instead of NO (NO uncoupling) (for review see ref. [7]). O2- eventually reacts with NO to form peroxinitrite, thus increasing vascular noxious agents and triggering a vicious cycle of vessel damage (Fig. 1). Interestingly, experiments in NOX-knockout animals provided some evidence that vascular NOX plays an important role in NOS uncoupling in humans [8]. NOS uncoupling has been observed in patients with endothelial dysfunction secondary to diabetes mellitus, hypercholesterolemia or essential hypertension [9-11] and may be present also in scleroderma patients in whom vascular abnormalities and endothelial cell injury are a widespread finding.
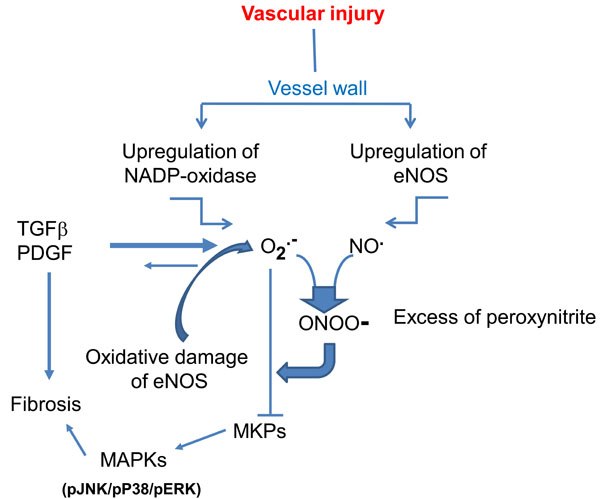
The vicious biochemical cycle that leads to excessive oxidative stress in vessel wall following persistent vascular injury, and to fibrosis (adapted from FÖrstermann and Münzel: Circulation 2006; 113: 1708-1714). Inactivation of MKPs (MAPK phosphatases) is responsible for activation of MAPKs.
Involvement of cyclooxygenase and lipoxygenase in ROS production have been demonstrated, but evidence of their participation in redox state and signaling is still scarce [12,13].
The most important enzymatic complex responsible for ROS generation is the membrane-bound NADPH oxidase (NOX). The prototypical phagocytic NADPH oxidase consists of two membrane subunits gp91phox and p22 phox, which form the flavoprotein known as cytochrome b558, and three cytosolic components p47phox, p67phox and the small G protein rac 2. Upon stimulation, p47phox is phosphorylated and forms a complex with the other cytosolic subunits. This complex migrates to the cell membrane where it combines with cytochrome b558 to constitute the active enzyme which transfers electrons from a substrate to O2, generating O2- [14]. The discovery of gp91phox homologues has allowed the identification of what is known as the Nox family of NADPH oxidase, which comprises seven members characterized by the catalytic subunits that they utilize. Thus, the family includes Nox1, Nox2 (formerly gp91phox), Nox 3, Nox4, Nox5, Duox1 and Duox2. All Nox family members are transmembrane proteins present in phagocytic and nonphagocytic cells. Nox1 has been detected in endothelial cells, vascular smooth muscle cells, colon epithelial cells. Nox2 is the catalytic subunit in phagocytes but is present in vascular, renal and neural cells. Nox3 is essential for normal vestibular function and its primary expression site is the inner ear. Nox4 can be found in vascular cells, fibroblasts and osteoclasts as well as in the kidney. Nox5 is found in testes and lymphoid tissue but also in vascular cells. Finally, Duox1 expressed in the thyroid gland, lung, pancreas, placenta, prostate and testis, Duox2 expression is more widespread [15].
Nox is activated in response to physical stimuli (shear stress, pressure), growth factors (platelet-derived growth factor, nerve growth factor, fibroblast growth factor, transforming growth factor β), cytokines (tumor necrosis factor, interleukin 1), metabolic factors (hyperglycemia, hyperinsulinemia, glycation end products, free fatty acids), G protein-coupled receptor agonists (serotonin, thrombin, endothelin, angiotensin II, bradykinin). In phagocytes the activation of Nox2 occurs when immune complexes interact with Fc or complement receptor.
In addition, hydroxyl radicals may be generated by an increase in iron, copper or other metals via a metal-catalyzed oxidation.
Finally enzymes such as myeloperoxidase of neuthophils and monocytes and eosinophil peroxidase can produce HOCl during bacterial phagocytosis.
However, high levels of intracellular ROS can also be generated by many environmental stimuli including ionizing radiations, smoke, environmental toxins, ultraviolet light, chemotherapeutics such as bleomycin, which promotes metal-catalyzed oxidation, or adriamycin, which undergoes auto-oxidation.
TARGETS OF ABNORMAL OXIDATIVE STRESS
At high concentrations ROS can damage cell structures, nucleic acids, lipids and proteins. The hydroxyl radical can react with all the components of DNA molecules, including the deoxyribose backbone, the purine and the pirimidine bases, e.g. with formation of 8-OH-G. Modification of genetic material may contribute to mutagenesis, carcinogenesis and ageing.
ROS actions on lipids may induce lipid peroxidation in cell membranes, with subsequent modification of membrane fluidity, loss of membrane protein functions, efflux of cytosolic components.
Some proteins are particularly sensitive to oxidation. Mitochondrial proteins, members of the ubiquitin-proteasome system, proteins involved in energy metabolism and chaperons are only some examples. Protein tyrosine phosphatases are relevant targets of ROS. As a result of excessive oxidative stress, they are reversibly inactivated. This leads to increased activity of tyrosine kinases, mitogen-activated protein kinases (MAPK), extracellular signal-regulated kinases (ERK) and c-jun amino-terminal kinase (JNK) secondary to reversible oxidative inactivation of cysteine-dependent serine/threonine phosphatases.
Additional effect of ROS is the reversible activation of both receptor- and non-receptor- tyrosine kinases such as the Src kinases and Janus kinase (JAK). Activated Src initiates MAPK, NF-kB and phosphoinositide 3-kinase (PI3K) signaling pathways.
Abnormal degradation of extracellular matrix may contribute to the pathogenesis of fibrotic disorders. The activity of matrix metalloproteinase enzymes (MMPs) and their inhibitors (tissue inhibitors of metalloproteinases, TIMPS) has been extensively investigated in idiopathic pulmonary fibrosis (IPF). Hayashi et al. have shown increased expression of MMP 2 and 9 and TIMPS 1 and 2 in the areas of alveolar damage in patients with IPF [16]. McKeown et al. found high levels of MMP 3, 7, 8 and 9 in bronchoalveolar lavage fluid from patients with IPF [17]. Work with experimental animal models led to similar results. The majority of MMPs are secreted as pro-enzymes and activated by proteolysis of a cystein-zinc active site termed “cysteine-switch”. ROS and RNS can modify the cysteine switch, with subsequent MMPs activation [18]. However, free radicals can inactivate proteases as well [18]. The timing of activation and inactivation of MMPs and the local concentrations of ROS are likely able to determine the entity of MMPs activation and the pathological outcome.
There is evidence that ROS accumulation can cause apoptotic cell death by release of apoptotic factors from damaged mitochondria. Mitochondrial glutathione can be depleted leading to mitochondrial membrane permeabilization, release of cytochrome c and death [19].
Caspases have a crucial role in apoptosis. Once activated by extrinsic or intrinsic pathways, they migrate to the nucleus causing the ultimate cell breakdown. The extrinsic pathway includes the Fas/Fas ligand system. The intrinsic pathway is due to internal cell stressors such as DNA damage, that activates pro-apoptotic proteins (Bax, Bac) and/or inhibits anti-apoptotic Bcl-2 proteins, leading to release of cytochrome c from mitochondria, activation of caspase 9 and, consequently, caspase 3 and 7 with cell apoptosis [20]. ROS and RNS may increase cell death by interacting with caspase 3 [21]. Alternatively, caspases can be transiently inhibited through reversible inactivation of the cysteine active-site. As a consequence, the resistance of fibroblasts to apoptosis may determine persistent matrix deposition. Thus, apoptotic and anti-apoptotic mechanisms may be induced by ROS/RNS and the biological consequences depend on the specific cell subtypes and organs involved in the process, and on the stage of the disease.
Nuclear transcription factors may also be a target of oxidative stress. Activator protein 1 (AP-1) activity is induced in response to certain metals in the presence of H2O2 (5). According to several reports, ROS are able to affect the activation of nuclear factor (NF)-kB. (NF-kB) is an inducible transcription factor involved in cell survival and differentiation, as well as in inflammation and cell growth [22]. The nuclear factor p53, which has the ability to stop cell cycle and initiate apoptosis upon cell damage, is activated in several conditions characterized by high oxidative stress, such as UV irradiation, hypoxia and gamma-radiation.
The transcription factor Hypoxia-Inducible Factor 1(HIF-1) has been identified as one of the main regulators of cellular adaptation to low oxygen tension. Under normal oxygen conditions, HIF-1α is hydroxylated by a prolyl hydroxylase and degraded upon binding of the von Hippel-Lindau protein, the protein Elongin C and by recruitment of an E3 ubiquitin-protein ligase [23]. Under hypoxic conditions, hydroxylase activity may be reduced either as a consequence of limited O2 availability or increased generation of ROS by mitochondria, which oxidize Fe (II) in the catalytic center of the enzyme, thereby rendering HIF-1α resistant to ubiquitination and degradation [24]. Accumulation of HIF-1 induces the expression of several genes essential for cell survival under hypoxic conditions.
In summary, many cell functions are influenced by oxidative stress. The nature and duration of stress, as well as the target cell types, account for the different biological consequences triggered by the excess of free radicals.
EVIDENCE OF OXIDATIVE STRESS IN SCLERODERMA
Recently, we have extensively reviewed the evidence of excessive oxidative stress in scleroderma [25] and its role in the pathophysiology of scleroderma [1]. Since then, other reports have supported our observations.
Ogawa and colleagues have found increased levels of total antioxidant power in 24% of scleroderma patients. This finding was interpreted as a global response to enhanced oxidative stress [26]. The same group suggested that antibodies against methionine sulfoxidereductase A (MSRA) may enhance oxidative stress by inhibiting the enzymatic activity of one of the antioxidant repair enzymes [27]. The biological consequences of abnormal generation of free radicals was investigated by Avouac J et al. Using an enzyme-linked immunosorbent assay, they determined the urinary levels of 8-idroxy-2’-dedeoxyguanosine (8-oxodG), a biomarker of endogenous oxidative damage to DNA, in 80 consecutive scleroderma patients and 39 controls. Higher levels were found in the scleroderma cohort [28]. This finding is in agreement with previous observations by Svegliati et al. who showed impressive damage of chromosomes with activation of DNA damage checkpoints, assayed by phosphorylation of histone H2AX and accumulation of p21 WAF. The chromosomal abnormalities were present in human skin fibroblasts in vivo before the primary cell cultures, and were continuously generated in culture, and amplified by ROS. In fact, the number of altered metaphases was significantly reduced by incubating the skin fibroblasts with ROS scavengers during the first and second day of culture [29]. Furthermore, the proteomic analysis performed by Aden et al. on skin biopsies of 12 patients with recent onset diffuse scleroderma, revealed abnormal abundance of proteins involved not only in extracellular matrix production, myofibroblast contractility, and energy metabolism, but also in response to oxidative stress [30].
Indirect proof of abnormal oxidative stress was provided by Dooley et al, who have shown that the antioxidant epigallocatechin-3-gallate can reduce extracellular matrix production and inhibit contraction of dermal fibroblasts from scleroderma patients. Furthermore, epigallocatechin-3-gallate was able to suppress intracellular ROS, ERK1-2 kinase signaling and NF-kB activity [31].
Studies in animal models of scleroderma have confirmed the relevance of oxidative stress in the pathogenesis of the fibrotic processes.
Abnormal NO metabolism has been demonstrated in the tight-skin (TSK-1/+) mouse model of fibrosis. As mentioned before, NO is a free radical synthesized from L arginine by NOS. Three main NOS isoforms have been identified. Neuronal and endothelial NOS (eNOS) are constitutively expressed in neuronal and endothelial cells, as well as in other cell types. An inducible isoform is expressed in response to a variety of stimuli. Expression of eNOS was significantly reduced in TSK-1/+ mouse, and decreased NOS activity was also detected in TSK-1/+ mouse tissue compared to wild type mice, suggesting an abnormal NO metabolism [32].
Skin and lung fibrosis induced in mice by administration of bleomycin is an experimental model of scleroderma exploited to investigate the molecular mechanisms leading to fibrosis. Interestingly, superoxide and hydroxyl radicals are generated following bleomycin administration [33]. Demonstration that antioxidant therapy may ameliorate lung injury and dermal fibrosis in bleomycin-treated mice [34-36] argues in support of oxidative stress influence in this experimental model. Mice devoid of p47 phox are deficient of free radical production through NADPH-oxidase. These mice exhibited reduced bleomycin–induced lung fibrosis [37]. Interestingly, a direct role of ROS in scleroderma has been suggested by Servettaz et al. [38]. They injected BALB/c SCID mice with several ROS-inducing agents and found that peroxynitrites induce skin fibrosis and serum anti-CENP-B autoantibodies as in limited cutaneous scleroderma. On the other hand, hypoclorite or hydroxyl radicals induced lung and skin fibrosis and DNA topoisomerase I autoantibodies, thus mimicking diffuse cutaneous scleroderma. In the latter mice, the ADAM/Notch signaling pathway appeared upregulated in the skin, lung, fibroblasts, and splenocytes as in the skin biopsy samples taken from control patients affected by scleroderma [39].
In summary, the data reported so far leave little doubt that free radical accumulation is a major event in the clinical history of a scleroderma patient, but the identification of the primary cause leading to ROS production is still a major challenge.
SOURCES OF FREE RADICALS INSCLERODERMA
Ischemia and reperfusion are a major cause of ROS production and lead to tissue damage. During ischemia, moderate ROS generation can be ascribed to mitochondria, whereas, upon reperfusion, the purine catabolites, accumulated as a consequence of exaggerated ATP consumption occurring during the ischemic phase, are metabolized, in presence of oxygen, by xanthine oxidase to yield massive amounts of superoxide and hydrogen peroxide. Neutrophils are the principal effector cells of such reperfusion injury.
Ischemia reperfusion events, clinically manifested in scleroderma patients as Raynaud’s phenomenon, have been considered the most likely contributors to the abnormal oxidative stress observed in this disorder. However, their contribution to the exaggerated oxidative stress of scleroderma is debatable for the following reasons: i) patients with primary Raynaud’s phenomenon failed to display direct or indirect evidence of abnormal oxidative stress [40]; ii) post-occlusive hyperemia was altered in scleroderma patients indicating a decreased reperfusion after ischemia [41]; iii) a clear inflammatory response such as that occurring after reperfusion is lacking in scleroderma patients; iv) un-stimulated monocytes from peripheral blood of scleroderma patients, rather than scleroderma neutrophils, release more superoxide in vitro [42]. However, it cannot be ruled out that chronic ischemia in scleroderma patients may be more important than single ischemic events.
In most cell types, ROS in scleroderma can also be the result of the interaction of cytokines or growth factors with their specific receptors, e.g. interleukin 6, interleukin 3, TNF-α, angiotensin II, PDGF, nerve growth factor, TGF-β1, fibroblast growth factor, granulocyte-macrophage colony-stimulating factor. This aspect has drawn increased attention over the last years and has allowed to clarify that ROS can function as second messengers in signaling cascade essential for cell proliferation and differentiation.
Of the several cytokines and growth factors implicated in the pathogenesis of scleroderma, it is worthwhile remembering that angiotensin II, PDGF and TGF-β1can interact with members of the NOX family in vascular smooth muscle cells, cardiac, lung and skin fibroblasts.
TGF-β is a potent profibrotic cytokine that plays a key role in the pathogenesis of scleroderma fibrosis. Increasing evidence suggests that this may occur via ROS generation. TGF-β increases ROS production in several non-phagocytic cells and in different cell compartments. Available information suggest that TGF-β stimulates ROS by interacting with a trans-membrane serine/threonine kinase coupled to NOX4 and modulating the function of mitochondria and microsomes [43,44]. Furthermore, TGF-β may increase ROS/RNS levels suppressing the expression of antioxidant enzymes, including catalase, superoxide dismutase and glutathione peroxidase [45], and by decreasing the intracellular concentration of glutathione [46]. Vascular endothelial growth factor (VEGF), angiotensin II and PDGF are also key players in scleroderma. They exert their pro-oxidative actions through receptors coupled to NOX4 (VEGF) and NOX1 and NOX 2 (Angiotensin II and PDGF) [47, 48].
Information regarding the distribution of NOX enzymes in scleroderma tissue and their interaction with different cell surface receptors are still limited, although the overproduction of free radicals in several scleroderma cell types suggests the link of these enzymes with cell surface receptors.
EFFECT OF OXIDATIVE STRESS ON SCLERODERMA FIBROSIS
Oxidants may directly activate fibroblasts or they may contribute to the establishment of fibrosis by altering the balance between protease and anti-protease (see above). The latter mechanism may be particularly active and pathophysiologically important in the lung, where disruption of the normal architecture and disorganization of the vasculature may create the basis for locally persistent ischemia-reperfusion events which may propagate the injury even in the absence of the triggering pathogenic insult.
Free radicals may also be operating through their link with the cytokines and growth factors implicated in the pathogenesis of scleroderma. Jobling et al. proposed that release of TGF-β from its latency association protein (LAP), a process indicated as TGF-β activation, which allows the binding of TGF-β to its receptor, is induced by oxidative modification of LAP at a redox switch located at methionine 253 [49]. In addition, free radicals can stimulate the expression and secretion of TGF-β in many cell types [50] and may mediate TGF-β-induced differentiation of fibroblasts to myofibroblasts [51], pro fibrotic activities [52] and epithelial-mesenchymal transition [53]. On the whole, there is evidence of a positive interaction between TGF-β and ROS. The same applies to PDGF. In fact, there are several proofs of the importance of the PDGF receptor-dependent pathway in the pathogenesis of fibrotic processes, of scleroderma in particular. Stimulatory auto-antibodies targeting PDGF receptor detected in serum from a cohort of scleroderma patients represent another source of free radical generation which can stimulate collagen and α-smooth muscle actin via Ras/Erk. Although the primary cause of these autoantibodies remains obscure, the persistent stimulation of PDGF receptor signaling in fibroblasts might account for increased collagen production and fibrosis in scleroderma [54].
OXIDATIVE STRESS AND FIBROSIS OF THE LUNG, LIVER AND KIDNEY
Several reports described the contribution of oxidative stress to the pathogenesis of fibrosis in organs such as the lung, the liver and the kidney.
Pulmonary fibrosis can be associated to connective tissue diseases or may be ascribed to radiation exposure, environmental agents such as asbestos and silica, or pharmacological agents such as bleomycin. When the triggering event causing the progressive fibrosis of the alveolar interstitium is unknown, the term idiopathic pulmonary fibrosis (IPF) is used [55].
The pathogenesis of IPF remains ill defined, but epithelial injury appears to be one of the initial steps followed by abnormal re-epithelization and deregulated remodelling of the extracellular matrix in a vain attempt to restore the integrity of the lung. Thus, many investigators consider IPF to be primarily a disease of abnormal wound healing.
Recently, a number of studies have suggested that ROS play a significant role in the progression of pulmonary fibrosis in animal models, and also in human subjects. In experimental animal models based on the administration of bleomycin, silica or asbestos, treatment with antioxidants such as N-acetylcysteine and desferoxamine confers a significant protection [56]. The same protection occurs in the presence of NADPH oxidase knockout or knockdown [57] or when extracellular superoxide dismutase is over-expressed [58]. In contrast, lack of superoxide dismutase worsens bleomycin-induced fibrosis [59]. With regard to human studies, increase of 8-isoprostane, a product of lipid peroxidation induced by free radicals, has been demonstrated in bronchoalveolar lavage fluid of patients with IPF [60], and in exhaled breath condensate [61]. Increase of other markers of oxidative stress, such as NO [62], hydrogen peroxide [61] and ethane [63], was shown in the exhale of IPF patients, and inversely correlated with glutathione levels in alveolar epithelial lining fluid [64] and with extracellular superoxide dismutase [65]. Daniil et al. reported that total hydroperoxides, a marker of oxidative burden, were higher in serum of IPF patients than in controls, and negatively correlated with lung function parameters [66]. In addition, lung specimens of IPF patients show elevated expression of iNOS [67]. Taken together, these findings suggest that an increased oxidative and nitrosative stress is a hallmark of IPF.
Major generators of free radicals in the lung of IPF patients are inflammatory cells. Neutrophils and eosinophilis in BAL are increased in 70-90% and 40-60% of IPF patients, respectively [68]. Their increase is associated with a worse prognosis and mortality [69]. Interestingly, Bocchinoetal have shown that IPF fibroblasts, like scleroderma fibroblasts, also contain large excess of ROS, due to activation of NADPH oxidase-like system, with higher levels of tyrosine phosphorylated proteins [70]. These traits tend to disappear with time in culture, as if they were dependent on a transient trigger. Oxidative stress in the lung can contribute to a persistent inflammation by activating NF-kB (nuclear factor-kappa B) and activator protein-1 (AP-1) [71,72], which in turn amplify the inflammatory milieu, the generation of oxidative stress, and the fibro-proliferative response. In this context, apoptosis of epithelial cells becomes prominent and elicits the vicious cycle of alveolar damage and abnormal repair.
Finally, ROS can directly damage components of the extracellular matrix including heparan sulphate and hyaluronic acid, through activation of matrix metalloproteinases and tissue inhibitors of MMP [73,74].
It is likely that some of the mechanisms responsible for the pathobiology and progression of fibrosis are shared by various clinical situations characterized by pulmonary fibrosis.
The impact of oxidative stress on lung function has prompted studies to correct the oxidant/antioxidant imbalance in IPF. In a randomised, double-blinded, placebo-controlled trial, oral N-acetylcysteine (NAC), a thiol-containing agent which promotes glutathione synthesis [75] and scavenges hydrogen peroxide, hydroxyl radical and hypochlorous acid, was added to the standard therapy (prednisone plus azathioprine) of IPF patients [76]. After one year of treatment, the addition of NAC resulted in a better preserved vital capacity and diffusing capacity for carbon monoxide. However, its limited therapeutic efficacy may be explained by the reduced bioavailability of the antioxidant after oral administration [77], by the toxic effect of the other drugs in the therapeutic regimen as suggested by G.W. Hunninghake [78], by the presence of an established lung damage that can be only partially reversed. With regard to scleroderma, few clinical studies involving small cohorts of patients have been undertaken to verify the clinical benefit of treatment aimed at reducing oxidative stress [79, 80].
Oxidative stress has also been proposed to be critical in the pathogenesis of liver fibrosis.
Chronic viral infections, long-term consumption of alcohol, ischemia-reperfusion events, metabolic disorders, and non-alcoholic steato-hepatitis can all induce cell damage through increased generation of ROS and growth factors by Kupffer cells and injured hepatocytes [81]. Hepatic stellate cells, portal fibroblasts, bone marrow derived mesenchymal stem cells and hepatocytes and biliary epithelial cells via epithelial to mesenchymal transition, once activated by ROS and profibrogenic soluble mediators such as TGF-β, PDGF, leptin and angiotensin II, are the main actors in the pathogenesis of liver fibrosis (for review see ref. [82]). Generation of ROS may occur via mitochondrial damage, mitochondrial electron transport chain, cytochrome P450 activation, NADPH oxidase and xanthine oxidase.
The molecular pathways triggered intracellular by ROS include NF-kB, JNK and activating protein-1 with a downstream upregulation of fibrosis related genes such as COL1A1, COL1A2, MCP1 and TIMP1 [82].
Further understanding of the mechanisms of ROS in the pathogenesis of liver fibrosis is, however, needed to pave the way to the successful development of novel therapeutic approaches.
Elevation of biomarkers of increased oxidative stress has been demonstrated in patients with chronic kidney disease, and tissue macrophages are a major source of reactive oxygen and reactive nitrogen species in renal fibrosis [83]. However, it remains to be clarified whether this is the result of the retention of oxidative products or may be ascribed to a failure of the kidney to reduce this oxidant products.
At the molecular level, angiotensin II is one of the main contributors to the progression of renal disease leading to the generation of ROS via NOX1 [84]. In this respect, it is worthwhile noting that angiotensin type 2 (AT2) receptor blockade decreased ROS generation in experimental models of chronic kidney disease, leading to amelioration of renal fibrosis [85].
Furthermore, a number of studies have indirectly supported the hypothesis that oxidative stress may contribute to the progression of renal fibrosis.
In experimental animals, diet-induced hypercholesterolemia is responsible for a more severe fibrosis, which is attributable to an increased deposition of oxidized lipoproteins in the renal interstitiumand in renal tubular cells, with subsequent macrophage infiltration and increased NF-kB signaling [86]. The deficiency of scavenger receptors CD36 and SR-A was responsible for less oxidized lipoprotein deposition and attenuated NF-kB activation [87,88].
Based on the clinical and experimental findings of increased oxidative stress in chronic kidney diseases, therapies designed to block scavenger receptors or antioxidants targeting specific oxidant steps have been tried with promising results. However, further studies are warranted.
CONCLUDING REMARKS
We have highlighted the important role that ROS/RNS have in the pathogenesis of scleroderma fibrosis.
We have recently proposed a comprehensive hypothesis which considers oxidative stress as one of the main culprits [1]. To summarize, an initial unknown event could trigger a slight increase of ROS in vessel walls. Persistent and/or higher activation of neighboring normal cells by permeable H2O2 may occur and generate a small nest of inflammatory cells releasing a large array of mediators. Inhibition of proteasome activity by low levels of ROS help stabilize proteins including Ras and ERK. The cell NADPH oxidase undergoes phosphorylation by ERK and stimulates the production of ROS which maintain the generation and amplification of ROS in a vicious circuitry. Free radicals, inflammatory cytokines and specific growth factors all contribute to the proliferation of vascular and perivascular cells. The main feature at this stage is vessel wall thickening with increasing hypoxia. Under hypoxic conditions, HIF-1 mediates a switch from oxidative to glycolytic metabolism which yields 18 times less ATP per mole of glucose consumed. In the presence of ATP, the NADPH-oxidase system is coupled to glutathione synthesis, but when less ATP is available the system is deregulated and less cellular glutathione is produced [89]. As a consequence, ROS cannot be buffered and a vicious cycle linking ROS, vascular abnormalities, extracellular matrix deposition and hypoxia is established. Fibrosis in skin and visceral organs dominates the clinical picture, with profound disruption of visceral organs architecture, which contributes further to tissue hypoxia.
This hypothesis would gain ground by solving the following issues:
- does ROS increase precede or follow scleroderma tissue lesions?
- are all clinical features of this disease dependent upon oxidative stress?
- how can ROS influence disease activity ? Novel animal models of scleroderma and studies of cohorts of patients at risk of developing scleroderma would probably help address these questions.
Another important point is how this pathogenic scenario might change our therapeutic approach. First, since fibrotic derangement of the lungs and visceral organs and microvascular abnormalities cannot be completely reversed once established, only an early treatment, hopefully based on targeting multiple molecules along this pathogenic signaling pathway, could prevent their formation. Second, taken into account that the activity of the distinct elements of the pathogenic scenario may vary from patient to patient, and even in the single patient at different stages of the disease, the identification of multiple biomarkers to reclassify patients according to disease activity is of paramount importance to treat the right patients with the right drugs combination.
Finally, the study of oxidative stress in lung, liver and kidney fibrosis can provide insights into the involvement of ROS/RNS in the pathogenesis of excessive extracellular matrix deposition in different compatments and may lead to the development of novel treatments potentially transferable to the therapy of scleroderma patients.
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
Declared none.

