All published articles of this journal are available on ScienceDirect.

The Role of the NLRP3 Inflammasome in Fibrosis
Abstract
Fibrosis leads to the deposition of collagens in organs and tissues. The resulting pathology induces a loss of function in the organ it is manifested in and this loss of function modulates the morbidity and mortality in that individual. Indeed, approximately 45% of all deaths in the Western world can be attributed to fibrosis and there are no FDA approved drugs for the treatment of fibrosis. The recent discovery of the inflammasome has led to a plethora of studies investigating this inflammatory signaling pathway in a wide variety of pathogen associated diseases. Many studies have focused on the NLRP3 inflammasome and this inflammasome is activated by a wide variety of cellular alarm signals. Once activated, caspase-1 is cleaved, inducing the secretion of IL-1β and IL-18 that signal to aid in the clearance of invading organisms. However, as the knowledge of the inflammasome has expanded, it was found that it can directly control collagen synthesis, leading to the increased deposition of collagens in the tissues such as the lung, liver, heart, and skin. Mice lacking the inflammasome adaptor protein, ASC, failed to become fibrotic when exposed to bleomycin. Inhibition of caspase-1 activity in fibroblasts from patients with the fibrotic disease systemic sclerosis, decreased collagen synthesis and reduced α-smooth muscle actin expression in myofibroblasts. Taken together, these observations suggest that the inflammasome can drive the fibrotic response and paves the way for novel therapeutics to be identified.
INTRODUCTION
Approximately 45% of all deaths in the Western world can be attributed to fibrotic diseases resulting from the increased accumulation of collagens in the internal organs or tissues [1]. Fibrosis is able to manifest in almost any organ that contains fibroblasts, including lungs, kidney, liver, and heart. It can affect a single organ or be systemic affecting numerous organs. The causative agent precipitating the development of fibrosis is often unknown; however, in some cases fibrosis can be attributed to pathogens [2,3] or inert substances such as radiation [4] and chemicals [5]. To date, there are no FDA approved drugs that control the accumulation of collagen in tissues.
THE INFLAMMASOMES
The inflammasomes are pattern recognition receptors that have recently been identified that are capable of recognizing a diverse range of conserved molecular motifs unique to microorganisms. In addition, they can detect chemical alarm signals produced by activated cells that are associated with host infection and tissue damage [6]. The main function of the inflammasome is to upregulate and activate the innate immune system via the secretion of IL-1β. There are different classes of pattern recognition receptors and all are evolutionary conserved e.g. toll like receptors, leucine rich repeats, and NOD-like receptors (NLRs). These receptors are involved in a wide variety of ligand sensing and in combination lead to the increased expression, and activation and secretion of IL-1β and IL-18. With the discovery of the inflammasome, it was found that leucine rich repeats contained within NLRs are a new class of pattern recognition receptor consisting of varying numbers of leucine repeats that allow the protein to fold into three dimensional curved structures [7,8]. These curved structures bind specific ligands found on pathogens [9]. NLRs are comprised of an N terminal effector domain, a central NOD or NACHT (a domain found in NAIP, CIITA, HET-E, and TP-1 proteins) and a ligand sensing C terminal leucine rich repeat domain [10]. Currently, twenty-three NLRs containing NOD and leucine rich repeat domains have been identified and have been sub-classified by their N terminal region; NLRC 1-5 contain a caspase recruitment domain (CARD) protein, NLRP 1-14 contain pyrin, and NAIPs contain a BIR protein [11]. An inflammasome is comprised of an NLR receptor that is the sensor. The NLR sensor is thought to detect the alarm signal. The mechanism of detection of that alarm signal is yet to be fully elucidated but once activated, this sensor induces the complex formation that is called the inflammasome. The inflammasome associates with an adaptor protein (e.g. apoptosis speck like protein containing a CARD, ASC) and also associates with pro-caspase-1. The resulting association of these proteins causes in the cleavage of caspase-1 and subsequent activation of that protein.
The NLRP3 inflammasome is one of the most extensively studied inflammasomes and is capable of sensing a wide variety of alarm signals [6]. Recently it was found that the assembly of the NLRP3 inflammasome requires the presence of reactive oxygen species [12]. The direct positional involvement of the endoplasmic reticulum (ER) and mitochondria was also shown to be very important in this signaling [13]. The role of the mitochondria in the innate immune response has been well characterized and has been reviewed by West et al., [14]. Quiescent NLRP3 is localized to ER structures; however, once the inflammasome is activated, both NLRP3 and ASC redistribute to the perinuclear region of the cell where they co-localize with the ER and mitochondria organelles [13]. These data suggests that reactive oxygen species and mitochondrial signaling can play a significant role in the assembly and activation of the NLRP3 inflammasome; however whether reactive oxygen species activates other inflammasome platforms is not known.
Expression of inflammasome proteins can be found in a wide variety of immune and non-immune cells, including monocytes/macrophages [15], T cells [16], myofibroblasts/fibroblasts [17], keratinocytes [18,19], epithelial cells [19,20], and hepatic stellate cells [21] further suggesting the importance of this inflammatory initiating pathway in the immune response.
CASPASE-1
The penultimate role for the assembly of the inflammasome is the cleavage and subsequent activation of caspase-1. Caspase-1 (also called IL-1β converting enzyme, ICE) is first translated as a 45 kDa inactive precursor [22] that contains a 15 kDa N-terminal subunit, a central 20 kDa subunit, and a 10 kDa C-terminal subunit [23]. The assembly of the inflammasome localizes other caspase-1 proteins together resulting in the autocleavage and activation of caspase-1 [23]. The active caspase-1 is comprised of a tetramer consisting of two 20 kDa fragments and two 10 kDa fragments [24]. Once activated, caspase-1 is then able to cleave a wide variety of protein precursors, inducing unconventional or leaderless protein secretion that occurs through an ER/Golgi-independent pathway [25,26]. Many of these proteins cleaved by caspase-1 have a role in the cytoskeleton of the cell [27,28], in glycolysis [27], mitochondria function [28], and inflammation [28]. In addition, once activated, caspase-1 induces its own secretion [28], perhaps in a mechanism that limits the cleavage of proteins by caspase-1.
IL-1β AND IL-18
The two most studied proteins cleaved by active caspase-1 resulting in their secretion from the cell containing an activated inflammasome are IL-1β and IL-18. IL-1β is processed as a 30.7 kDa inactive protein that is cleaved to the active 17.5 kDa protein. Likewise, IL-18 is initially processed as a larger 22.3 kDa inactive protein that is cleaved by caspase-1 to a 17.3 kDa active form. Once cleaved by caspase-1, both IL-1β and IL-18 are secreted from the cell where they can be involved in autocrine and paracrine signaling. IL-1β and IL-18 are structurally similar and are some of the few proteins known to contain only β-pleated sheets [29].
IL-1β is a pleiotropic cytokine that is involved in localized inflammation targeting parasitic, bacterial, or viral infections; or involved in systemic inflammation as a result of chronic disease. IL-1β is produced by many cell types e.g. epithelial cells, fibroblasts, T cells, etc; in response to pathogens or damaged tissues. IL-1β can modulate the expression of its own mRNA via signaling mediated by the IL-1 receptor and the signaling pathways involved induce increased p38 MAPK phosphorylation and NFκB activity [30]. In cells stimulated with IL-1β, the mRNA levels are sustained for over 24 hours [30], whereas in cells stimulated with lipopolysaccharide or phorbol 12-myristate 13-acetate, IL-1β mRNA levels are significantly reduced by 6 hours [31]. IL-1β requires two signals for the secretion of the protein; the first signal usually obtained via toll-like receptor signaling or IL-1 receptor signaling upregulates IL-1β gene transcription and the second signal is obtained via inflammasome activation, cleaving caspase-1 leading to IL-1β maturation and secretion. The acute release of IL-1β into the peripheral blood in response to cellular stimuli upregulates the expression of other proinflammatory cytokines such as IL-6; it also raises cortisol levels, and acts on the hypothalamus to induce fever. However in some chronic diseases, the amount of IL-1β that is secreted may not be enough to stimulate the hypothalamus and may only act locally to enhance inflammatory gene expression leading to chronic disease.
The function of IL-18 (previously called interferon-γ inducing factor) also participates in downstream inflammasome signaling but the effects of this cytokine are yet to be fully elucidated. Unlike IL-1β, IL-18 is constitutively expressed in its precursor form in many cells and only requires caspase-1 activation for its cleavage and secretion. IL-18 can also be cleaved by proteinase 3 [32], whereas cleavage by caspase-3 leads to inactivation of this protein [33]. IL-1β does not appear to be cleaved by caspase-3 or proteinase 3. Like IL-1β, IL-18 can modulate the expression of its own RNA and its signal transduction is similar to that of IL-1β via NFκB activity [29]. IL-18 can activate T cells resulting in the increased synthesis of TNF-α, IL-2, and GM-CSF and polarizes the Th1 response by suppressing IL-4 [29].
IL-1β AND IL-18 CAN INDUCE FIBROSIS
The initiating molecular mechanisms that are involved in acute and chronic disease are yet to be fully elucidated; however IL-1β and IL-18 appear to be central to both pathologies. What determines whether an acute response to an initiating event with resolution of disease versus a chronic response leading to unresolved disease and fibrosis is still unknown. However, recent studies have demonstrated a dichotomous signaling role by IL-1β and its effects on TGF-β1 expression. In studies by Luo et al. [34], they demonstrate that a short stimulation (minutes) with IL-1β and TGF-β1 resulted in the inhibition of Smad3 phosphorylation and further inhibited downstream TGF-β1 signaling pathways. However, a long exposure (24 hours) with IL-1β and TGF-β1 increased Smad3 phosphorylation [34] and induced TGF-β1 expression. In addition, it has been shown that long exposure of microvascular endothelial cells to IL-1β can induce the permanent transformation of these cells to myofibroblasts [35]; and once differentiated, myofibroblasts synthesize large amounts of collagen.
The role of IL-1β in fibrosis and wound healing is now becoming significantly apparent. In vitro studies demonstrate that IL-1β (and IL-1α) can stimulate collagen expression in a dose dependent manner [36]. Transient overexpression of IL-1β in airway epithelial cells promoted the release of TNF-α and IL-6, followed by a significant increase in TGF-β1 and platelet derived growth factor [37] that induced the deposition of collagen in the lung. Because IL-1β can induce its own gene expression, chronic activation of the inflammasome, resulting in the continual cleavage of IL-1β in a positive feedback mechanism could conceivably maintain an elevated level of active TGF-β1 protein resulting in fibrosis.
To further elucidate the role of IL-1β in the development of fibrosis, an investigation of deep incisional wound healing in mice deficient for IL-1 receptor. Thomay, et al. [38], reported decreased fibrosis, more collagenolytic activity without a decrease in inflammatory cell infiltrates and also reported improved wound healing. More importantly, the tensile strength of the wound was not impaired in this model. In addition, wound fluids contained less IL-6, TGF-β1, and vascular endothelial growth factor. In addition, the administration of Anakinra (IL-1 receptor antagonist) to the deep wounds abrogated wound fibrosis [38] further suggesting that IL-1 signaling in this setting is profibrotic.
IL-18 has also been shown to be profibrotic. Administration of recombinant IL-18 induced myocardial remodeling and increased interstitial myocardial fibrosis [39], whereas the direct targeting of IL-18 by the compound Felodipine reduced perivascular fibrosis [40]. The mode of action of Felodipine is to block calcium channels. Calcium channel signaling has recently been shown to be important for inflammasome activation [41,42]; therefore, the reduced secretion of IL-18 may in fact be directly due to the inhibition of the inflammasome rather than the direct targeting of Felodipine to IL-18.
In vitro studies demonstrate a dose dependent increase in type I and III collagen proteins with recombinant IL-18 and increased collagen gel contraction [43]. IL-18 was also shown to induce fibroblast proliferation [43] and interestingly, IL-18 induced α2β1 integrin [43]. This integrin is one of the major receptors that bind collagens. Other profibrotic molecules have been shown to be increased with the administration of recombinant IL-18 e.g. IL-18 promoted increased osteopontin expression and this was found to coincide with increased in cardiac collagen content [44].
NLRP3 INFLAMMASOME AND FIBROSIS
The involvement of the inflammasome in fibrosis is slowly being elucidated. The conundrum is that the inflammasome appears to be involved in many innate responses to pathogens or cellular alarm signals; and yet, fibrosis does not result in every case. In fact, fibrosis seems to be the exception rather than the rule. This suggests that fibrosis resulting from activated inflammasome(s) could be dependent on numerous factors. These factors could include the type of inflammasome(s) that are activated; genetic variation(s) that affect the autocrine or paracrine responses by cells to the inflammasome-mediated signalosome; the cell type that the inflammasome is activated in; or possibly, by a combination of all these factors.
Activated fibroblasts (myofibroblasts) not only produce excessive amounts of collagen when the skin is breached they are also able to present antigens [45]. Historically, fibroblasts have not been considered central to the immune response, however myofibroblasts from different organs have antigen presenting capabilities [46,47]. What is now becoming apparent is that fibroblasts can function as sentinel cells, capable of becoming activated by bacterial products (or other alarm signals) and once activated secrete cytokines and chemokines [48]. Furthermore, when activated, they have a functional inflammasome [17]. Under normal wound healing conditions, myofibroblast differentiation and activation results in the synthesis of high levels of collagen. Once the wound is closed, myofibroblasts usually undergo apoptosis; however, when this mechanism becomes dysregulated chronically activated myofibroblasts can cause progressive fibrosis.
Gasse et al., [49] were the first to investigate the connection between the inflammasome, the IL-1 receptor, and MyD88 signaling with fibrosis. Mice that were deficient in the ASC protein had attenuated responses to the profibrotic compound, bleomycin. This extensive study demonstrated the integral role of IL-1 signaling via its receptor (IL-1R1) and MyD88 in pulmonary fibrosis. In mice that were IL-1R1 deficient and those that were MyD88 deficient had an abrogated responses to bleomycin. Furthermore, the direct administration of recombinant mouse IL-1β to the lungs of wild-type mice mediated a marked increase in tissue destruction with inflammation and collagen deposition. The addition of IL-1 receptor antagonist appeared to be more effective at limiting fibrosis in the wild-types than the administration of IL-1β neutralizing antibodies. IL-1β production was found to be more dependent on the ASC inflammasome signaling rather than toll-like receptor signaling [49].
Recently, it was found that uric acid is released into the lungs when cells are stimulated with bleomycin [50] and can act as a danger signal. It is thought that the localized increase in uric acid results in the deposition of crystals inducing cell membrane damage causing activation of NLRP3, and release of IL-1β. Gasse et al., [50] demonstrated that inflammatory signaling mediated by uric acid was dependent on IL-1 receptor, MyD88 and the NLRP3 inflammasome, and that this could be an autocrine loop mediating fibrosis. They found that caspase-1 deficient mice had an attenuated responses to bleomycin. They also demonstrate that there is some cross-talk between TLR2 and TLR4 as TLR2 and TLR4 deficient mice had abrogated responses to uric acid.
Studies on elastase-induced emphysema by this same group, also demonstrated that alveolar breakdown and subsequent fibrosis in the lung was dependent on ASC inflammasome-mediated signaling that released IL-1β. This finding could be attenuated with the IL-1 receptor antagonist, Anakinra [51]. Furthermore, Couillin et al., [51] demonstrated that the administration of elastase caused the dying cells to released uric acid. The subsequent addition of uric acid crystals to murine airways resulted in a transient increase in tissue inhibitor of metalloproteinases [50], a protein that inhibits extracellular matrix degradation. Importantly, the release of IL-1β in this model was not found to be cause by elastase cleavage of pro-IL-1β but was mediated via the activity of caspase-1 [51].
Silicosis has also been reported to be driven by the NLRP3 inflammasome resulting in the excessive deposition of collagen and recruitment of inflammatory cells to the damaged tissues [52]. Cassel et al., [52] found that the silica crystals induced reactive oxygen species in macrophages. In elegant studies, they found that by blocking the generation of reactive oxygen species this inhibited the activation of caspase-1, and the subsequent release of IL-1β and collagen synthesis. These studies further confirm the role of reactive oxygen species in NLRP3 inflammasome signaling and suggest that reactive oxygen species may promote a profibrotic phenotype.
We recently reported the role of the inflammasome in the fibrotic disease systemic sclerosis (SSc) [17] that mediated the increased secretion of collagen and induced myofibroblast differentiation. In a study investigating inflammasome gene expression and downstream inflammasome-mediated signaling products, we identified 40 genes that were upregulated greater than 2-fold. Several of these genes were found to be inflammasome platforms e.g. AIM2 and NLRP3. AIM2 is a sensor for cellular microbial dsDNA [53] and NLRP3 is a general sensor for cellular stress [54]. Furthermore, caspase-1 activity was upregulated approximately 2-fold in SSc fibroblasts and we found that fibroblasts isolated from the fibrotic lesions secreted more IL-1β and IL-18 protein and that this secretion could be abrogated with chemical or siRNA inhibition of caspase-1 [17]. This data suggests that the release of IL-1β and IL-18 is mediated by an inflammasome activation mechanism that is driving fibrosis. To test this hypothesis we inhibited caspase-1 and observed a significant reduction in the secretion of hydroxyproline (total collagen) by SSc fibroblasts [17]. The pathogenic cell that drives the increased collagen synthesis in the skin and organs is the myofibroblast. Inhibition of caspase-1 (chemically and with siRNA) reduced the expression of α-smooth muscle actin. Specifically, we observed that α-smooth muscle actin stress fibers contained less protein and were thinner in diameter in myofibroblasts treated with caspase-1 inhibition, whereas f-actin remain unchanged [17]. Furthermore, endogenous levels of α-smooth muscle protein and f-actin in quiescent fibroblasts remained unaltered. We confirmed the role of the inflammasome in fibrosis and found that ASC deficient and NLRP3 deficient mice had abrogated collagen deposition in the dermis when injected subcutaneously with bleomycin. Furthermore, as bleomycin induces circulating IL-1β and IL-18 we did not observe pulmonary fibrosis in the lungs of these mice, whereas the wild-type mice had increased deposition of collagen in the lung. In SSc, fibrosis directly correlates with morbidity and mortality. Our findings implicate that caspase-1 activity is directly involved in SSc fibrosis and suggests that there may be autocrine signaling mediated by IL-1β and/or IL-18 that promotes the profibrotic phenotype in these patients. The etiology of SSc is elusive and chemicals and pathogens have been previously reported in the pathogenesis of this disease [55, 56]. One hypothesis is that oxidative stress precedes fibrosis in SSc tissues further implicating the role for the inflammasome in disease pathogenesis [57].
It is now becoming apparent that other fibrotic diseases can be mediated by NLRP3 inflammasome signaling. The liver is easily injured by viruses and chemicals and responds to these insults by establishing inflammation that can lead to fibrosis. Like other tissues, fibrosis in the liver is a result of excessive collagen production accompanied by reduced extracellular matrix degradation. Upon activation, hepatic stellate cells differentiate into myofibroblasts and upregulate collagen secretion. Hepatic stellate cells are very similar to the type of myofibroblasts that are found in injured skin and are able to phagocytose, present antigen, contain α-smooth muscle actin stress fibers, and are capable of migration [58]. Thus, the finding that monosodium urate crystals could also activate the inflammasome in hepatic stellate cells resulting in the increase in collagen deposition in the liver is further suggestive that this important signaling pathway could be central to fibrotic diseases. In a study by Watanabe et al. [21], they were able to demonstrate that the liver antagonist monosodium urate upregulated TGF-β1 and type I collagen, induced the reorganization of actin, and promoted cellular stellation. These features were abrogated in NLRP3 and ASC deficient mice. Further support to these findings has led to the report by Gieling et al. [59], who showed that IL-1 mediates the progression of liver fibrosis. They demonstrate that on injury with thioacetamide, IL-1α and IL-1β peak on day 1 followed by a peak in type I collagen on day 3. They also demonstrate the increase in α-smooth muscle actin indicative of myofibroblast differentiation.
Further evidence that the inflammasome can promote fibrosis was demonstrated in a study on cardiac fibroblasts [60]. These studies support the finding that hypoxia with reoxygenation mediated by reactive oxygen species and potassium efflux stimulated the inflammasome in cardiac fibroblasts but did not stimulate the inflammasome in cardiomyocytes. Because cardiac fibroblasts can proliferate in the heart and produce extracellular matrix proteins, cytokine and growth factors, this investigation underlies the fact that these cells can act as sentinel cells able to sense danger signals resulting from ischemia/reperfusion to enhance the inflammatory response in the heart inducing deposition of collagens [60]. In ASC deficient mice, there was attenuated numbers of infiltrating macrophages and neutrophils in response to the ischemia.
SUMMARY
Taken together, these studies on fibroblasts and organs (lung, skin, heart, and liver); have demonstrate that the NLRP3 inflammasome can orchestrate the development of fibrosis in both sterile conditions and in pathogen driven models. In a fibrotic setting, a chronically activated inflammasome mediating the continuous release of IL-1β and IL-18, that induces autocrine signaling in fibroblasts could conceivably maintained the profibrotic phenotype in fibroblasts (Fig. 1). Whether fibroblasts require the activation of the NLRP3 inflammasome only, or require activation of other inflammasomes is yet to be determined. What is apparent is the cooperative regulation between TGF-β1 and IL-1β in maintaining an autocrine signaling loop mediated through the ERK1/2 signaling pathways [61] that is most likely operational in any cell capable of expressing collagens. Furthermore, TGF-β1 and IL-1β exhibited synergistic effects in promoting collagen stiffness [62] that may further promote the secretion of additional collagens into the extracellular space and/or the additional release of TGF-β1 [63]. Understanding the role of the NLRP3 inflammasome in chronic diseases that promote collagen synthesis will elucidate novel targets for therapy for this spectrum of diseases that to date, are difficult to treat. One of these therapies could be Colchicine as retroperitoneal fibrosis responds well to Colchicine therapy [64].
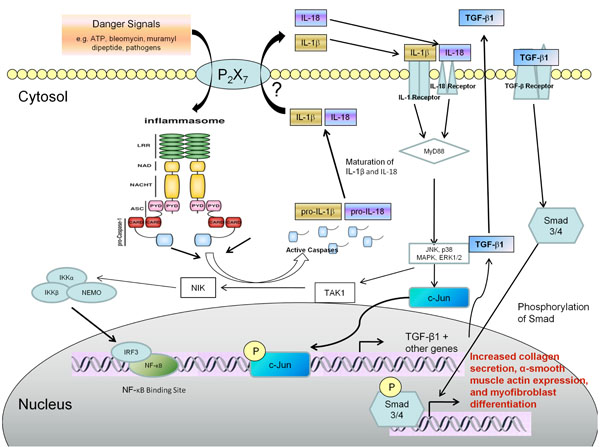
Autocrine signaling of IL-1β or IL-18 mediated by an activated inflammasome could promote myofibroblast differentiation and collagen deposition leading to fibrosis. Danger signals such as bleomycin, muramyl dipeptide, and various pathogen-related danger signals induce the assembly of the inflammasome. Once assembled the inflammasome mediates the cleavage of caspase-1 into its active form. Caspase-1 is then able to proteolytically cleave pro-IL-1β and pro-IL-18 to their mature active forms and these cytokines are secreted from the cell via a mechanism that is not fully understood. Both active IL-1β and IL-18 can autocrine signal in the fibroblast to upregulate p38 MAPK, JNK, and ERK1/2 signaling pathways resulting in increased gene expression e.g. TGF-β1 and other profibrotic genes. Active TGF-β1 is secreted from the cell and this in turn could autocrine signal to stimulate further collagen synthesis and increased α-smooth muscle actin incorporation into stress fibers that are indicative of myofibroblast differentiation.
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
The author has no conflict of interest.

