All published articles of this journal are available on ScienceDirect.

Caveolin-1 Signaling in Lung Fibrosis
Abstract
Caveolin-1 is a master regulator of several signaling cascades because it is able to bind to and thereby inhibit members of a variety of kinase families. While associated with caveolae and involved in their generation, caveolin-1 is also present at other sites. A variety of studies have suggested that caveolin-1 may be a useful therapeutic target in fibrotic diseases of the lung and other tissues because in these diseases a low level of caveolin-1 expression is associated with a high level of collagen expression and fibrosis. Reduced caveolin-1 expression is observed not only in the fibroblasts that secrete collagen, but also in epithelial cells and monocytes. This is intriguing because both epithelial cells and monocytes have been suggested to be precursors of fibroblasts. Likely downstream effects of loss of caveolin-1 in fibrosis include activation of TGF-β signaling and upregulation of CXCR4 in monocytes resulting in their enhanced migration into damaged tissue where its ligand CXCL12 is produced. Finally, it may be possible to target caveolin-1 in fibrotic diseases without the use of gene therapy. A caveolin-1 peptide (caveolin-1 scaffolding domain) has been identified that retains the function of the full-length molecule to inhibit kinases and that can be modified by addition of the Antennapedia internalization sequence to allow it to enter cells both in vitro and in vivo.
SUBCELLULAR AND CELLULAR LOCALIZATION OF CAVEOLIN-1
Caveolae are flask-shaped invaginations in the plasma membranes of cells that function in vesicular trafficking and in the compartmentalization of specific signaling cascades [1]. While caveolin-1 is well know as a molecule that is associated with caveolae and regulates their formation, there is an extensive literature demonstrating the association of caveolin-1 with other cellular organelles, the trafficking of caveolin-1 from caveolae to other organelles [2, 3], and even cell-surface [4] and soluble caveolin-1 [1]. At present, there is little or no evidence to suggest whether all sites of expression of caveolin-1 or only some sites (e.g. caveolae) are sites of action for caveolin-1 in the regulation of fibrosis.
Caveolin-1 is highly expressed in a variety of cell types in solid tissues including epithelial cells, endothelial cells, fibroblasts, and adipocytes. Fibroblasts are of obvious importance in fibrosis because they are the cells that overexpress collagen. Epithelial and endothelial cells are important because one source of fibroblasts (see below) is cells that have undergone epithelial-mesenchymal or endothelial-mesenchymal transformation. Endothelial cells are also important because of their roles in angiogenesis and inflammation and the importance of these processes in fibrosis. There are two types of epithelial cells in the lung, type I pneumocytes and the highly secretory type II pneumocytes. While it is agreed that caveolin-1 is expressed at high levels by type I pneumocytes, whether caveolin-1 is also expressed by the type II pneumocytes is a controversial topic. We observed prominent caveolin-1 staining in type II pneumocytes [5] and other images in the literature also appear to show type II pneumocyte staining [6]. It is possible that the idea that type II pneumocyte do not express caveolin-1 comes from in vitro experiments in which the cells may lose caveolin-1 in culture.
Caveolin-1 is also present in various classes of leucocytes including monocytes/ macrophages, PMNs, mast cells, and lymphocytes and regulates a variety of the immune functions of these cells [7-12]. These cell types are important in fibrosis because of their participation in inflammation, a process that is intertwined with fibrosis in that tissue damage leads to inflammation which leads to the expression of cytokines, chemokines, and growth factors that promote myofibroblast differentiation and their overproduction of ECM proteins [13] and fibrocyte recruitment into damaged tissue [14, 15]. In turn, these processes lead to further rounds of tissue damage and inflammation. In addition, monocytes are important in fibrosis because they serve as precursors to fibrocytes (see below) which in turn differentiate into fibroblasts and/or secrete factors that promote the proliferation and activation of fibroblasts.
PERTURBATION OF CAVEOLIN-1 FUNCTION
Investigators of caveolin-1 function and signaling have available to them the usual reagents: siRNA to inhibit expression, virus for overexpression, and knockout mice. In addition, the field has benefited greatly from knowledge of a peptide known as the caveolin-1 scaffolding domain (CSD, amino acids 82-101 of caveolin-1) which is the site within caveolin-1 where its function in inhibiting several kinases is localized [16-19]. When synthesized as a fusion peptide on the C terminus of the Antennapedia internalization sequence [20] or when lipidated [21], CSD can enter cells. CSD attached to the Antennapedia internalization sequence (Fig. 1) is sometimes referred to as cavtratin [22]. Henceforth we will simply refer to CSD attached to the Antennapedia internalization sequence as CSD. Like the intact molecule, CSD binds to kinases and inhibits their activity [21-24]. CSD is particularly useful because it is functional when delivered in vivo [20, 21, 25].
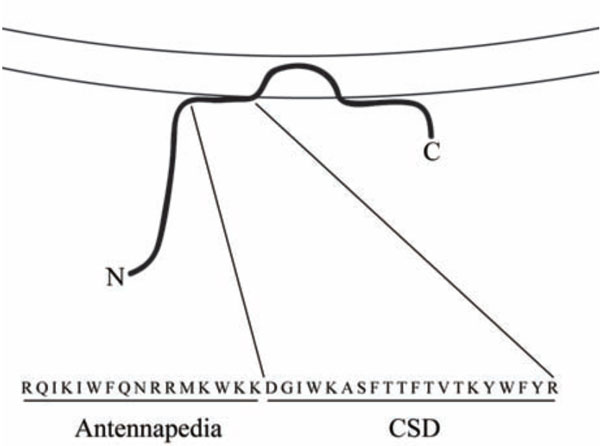
Structure of CSD. The association of caveolin-1 (thick black line) with membranes (parallel lines) is shown. Note that CSD is found at the surface of the membrane about midway between the N-terminal (N) and C-terminal (C) ends of caveolin-1. The combined amino acid sequence of the Antennapedia internalization sequence (Antennapedia) which carries CSD across membranes and into cells and of CSD is shown in single-letter code.

The Caveolin-1/CXCR4/CXCL12 Axis in Fibrosis. Profibrotic signals induce the differentiation in the bone marrow of a population of CD45+/CXCR4+/ColI+/caveolin-1 low cells that can be either CD14+ (filled circles) or CD14- (open circles). Under the influence of CXCL12, these cells traffic into the blood, then into damaged lung tissue where they begin to lose CD45 and to differentiate into myofibroblasts. CD45+/CXCR4+/ColI+/CD14- cells are fibrocytes, CD45+/CXCR4+/ColI+/CD14+ cells are referred to as collagen-producing monocytes [70].
The identification of direct targets for the CSD peptide (i.e. the kinases to which it binds directly) is a difficult question. While the list of signaling proteins that co-immunoprecipitate with caveolin-1 and/or whose activity is inhibited by CSD is long [26], many of these proteins may be indirect targets that are complexed with direct targets. To determine whether a protein is a direct target for the CSD peptide, it is necessary to study the interaction between the purified protein and the CSD peptide. The list of proteins shown in this manner to be directly affected by the CSD peptide includes eNOS, PKC, MEK, ERK, Ras, G proteins, and the EGF receptor [16, 27-30]. In 1997, in a phage display experiment the sequences ФXФXXXXФ and ФXXXXФXXФ (and the combined sequence ФXФXXXXФXXФ) where Ф stands for any of the aromatic amino acids (F, W, or Y) and X stands for any amino acid were identified as consensus CSD ligands [16]. At least one of these caveolin-1-binding domain (CBD) sequences is present in almost every protein known to interact with caveolin-1 [16, 31]. This definition of the CBD domain may be overly stringent. G proteins and G protein-coupled receptor kinases bind to the CSD peptide via sequences in which a hydrophobic amino acid (I, V, or L) is substituted for an aromatic amino acid at certain positions in consensus CBD sequences [16, 32]. These more inclusive consensus CBD sequences are ФXZXXXXФ and ZXXXXФXXZ where Z stands for F, W, Y, I, V, or L.
To address whether MAP kinases, PI3 kinases, and Akt may be direct CSD ligands, we compiled a table of the number of “classic” and “inclusive” consensus CBD domains in these molecules [25]. While none of the MAP kinases (including ERK and JNK) contain classic CBD sequences, several MAP kinase kinases (including the ERK kinases MEK1 and MEK2 and the JNK kinase MP2K7) and several MAP kinase kinase kinases and kinase kinase kinase kinases contain classic CBD sequences. However, if inclusive consensus CBD sequences are considered, then every MAP kinase of each degree contains multiple CBD sequences and thus may be a direct CSD target. Similarly, the use of inclusive sequences rather than classic sequences highlights PI3 kinases and Akt as direct targets for CSD.
Another novel resource for studying caveolin-1 function and signaling is caveolin-1 knockout mice in which caveolin-1 has been re-introduced into endothelial cells under the control of the preproendothelin-1 promoter [33]. In caveolin-1 KO mice, fibrosis (measured in terms of collagen expression) is increased in both the heart and the lungs [34]. Interestingly, in caveolin-1 RC mice, the overexpression of collagen is reversed in the heart, but is only slightly decreased in the lungs as compared to caveolin-1 KO mice. This observation suggests that fibrosis in the heart is regulated differently than fibrosis in the lungs at the cellular and/or molecular levels.
FIBROSIS OVERVIEW
The cell type most frequently associated with fibrosis is the myofibroblast. Myofibroblasts are fibroblasts that exhibit certain muscle-like properties: they are contractile and contain the marker protein α-smooth muscle actin (ASMA) [35]. They secrete high levels of ECM proteins and thus are key participants in the tissue remodeling that occurs during wound healing and in fibrotic diseases [36, 37]. In contrast, there are many more myofibroblasts in fibrotic lung tissue, particularly at sites of collagen deposition. Originally, myofibroblasts were viewed as resident fibroblasts that became activated and proliferated due to their interaction with effector molecules present in fibrotic lung tissue, such as thrombin and TGFβ. More recently, it has been proven that myofibroblasts can be generated by epithelial-mesenchymal and endothelial-mesenchymal transformation [31, 38, 39] and by the differentiation of bone marrow-derived stem cells into monocytes that further differentiate into circulating fibrocytes that express both fibroblast markers (collagen, spindle shape in culture) and hematopoetic cell markers (CD45, CD34, CXCR4). Fibrocytes then traffic into injured lung tissue where they differentiate into myofibroblasts [40-42]. In addition, monocytes and fibrocytes contribute to tissue remodeling and fibrosis by producing high levels of cytokines, fibrogenic growth factors, and MMPs [14, 41, 43-45]. Fibrocytes have been proposed to be essential participants in fibrotic diseases in several tissues and key targets for novel treatments for these diseases [46-50].
It remains to be determined whether myofibroblasts are derived primarily from resident fibroblasts, from fibrocytes, or by epithelial-mesenchymal transformation. Nevertheless, it has been elegantly demonstrated in irradiated mice reconstituted with fluorescent bone marrow cells that a major portion of the fibroblasts present in bleomycin-treated lung tissue are derived from fibrocytes [40, 41, 51, 52]. These observations indicate that inflammation and fibrosis are linked in two ways: 1) inflammatory cells secrete cytokines that promote the expression of collagen and other ECM proteins by fibroblasts, and 2) inflammatory cells (i.e. fibrocytes) serve as progenitors for a major portion of the activated fibroblasts in fibrotic lung tissue.
CAVEOLIN-1 IN LUNG FIBROSIS - FIBROBLASTS
Fibrosis-related diseases such as scleroderma (SSc) and idiopathic pulmonary fibrosis (IPF) are characterized by increased deposition of collagen and other extracellular matrix (ECM) proteins [53]. The major cause of morbidity and mortality in scleroderma is pulmonary fibrosis [54, 55]. Activated fibroblasts (known as myofibroblasts) are not present in normal lung tissue, but are found at sites undergoing active deposition of ECM proteins in fibrotic lung tissue. Moreover, pulmonary fibrosis may spread within the lung due to the apoptosis of epithelial cells induced by factors secreted by myofibroblasts [36, 56].
We and others have linked caveolin-1 to the regulation of fibrosis through its effects on the regulation of ECM production by fibroblasts. High levels of caveolin-1 are found in normal lung fibroblasts (NLF), whereas much lower levels are found in lung fibroblasts isolated from the fibrotic lung tissue of scleroderma patients (SLF) and IPF patients [5, 25, 57]. The loss of caveolin-1 results in the hyperactivation of signaling molecules (MEK, ERK, JNK, Akt) leading to the overexpression of collagen, tenascin-C, and the myofibroblast marker ASMA. When caveolin-1 expression is inhibited using siRNA, collagen expression and signaling molecule hyperactivation increase dramatically [5]; when caveolin-1 function or expression is enhanced using CSD or adenovirus, collagen expression and signaling molecule activation are inhibited [25, 57]. While CSD affects molecular events downstream from caveolin-1, it does not affect the expression of caveolin-1 itself. It does, however, affect the subcellular distribution of caveolin-1 [25], presumably because it interferes with the interaction of caveolin-1 with other proteins. We also found that caveolin-1 levels in lung fibroblasts could be regulated by PKCα, but not PKCε [5]. Transfection of these cells with constitutively active PKCα promoted caveolin-1 expression while transfection with dominant negative PKCα inhibited caveolin-1 expression.
In addition to regulating signaling molecule activation, caveolin-1 can regulate signaling molecule expression. PTEN (a dual lipid/protein phosphatase that negatively regulates proliferation by repressing the integrin-PI3K/Akt pathway), like caveolin-1, is present at reduced levels in IPF fibroblasts [58]. The lack of PTEN results in the activation of PI3K/Akt signaling. Caveolin-1 overexpression restores PTEN expression in these cells and caveolin-1 and PTEN also directly interact. Therefore, the decreased caveolin-1 expression observed in IPF fibroblasts facilitates their hyperproliferation, probably by allowing the cells to circumvent the suppression of proliferation initiated by the binding of polymerized type I collagen to its integrin receptor.
Two groups observed that in caveolin-1 null mice lung tissue shows significant pathology: the diameter of alveolar spaces is reduced, alveolar walls are thickened and hypercellular, and ECM deposition is significantly increased [34, 59]. Similarly, caveolin-1 levels are strikingly decreased in lung tissue induced to become fibrotic by irradiation or by bleomycin treatment [5, 60] and in fibrotic lung tissue from human scleroderma and IPF patients [25, 57]. Finally, intratracheal administration of adenovirus mediating the overexpression of caveolin-1 [57] or systemic administration of the CSD peptide [25] blocks the progression of bleomycin-induced lung fibrosis in mice. Thus, there is a clearcut causal relationship between low caveolin-1 levels and lung fibrosis.
In vivo experiments suggest that the same signaling cascades that regulate the expression of collagen, tenascin-C, and ASMA in NLF and SLF also regulate their expression in the fibrotic lung tissue of scleroderma patients and in the fibrotic lung tissue of mice treated with bleomycin. In particular, in the lung tissue of scleroderma patients and of bleomycin-treated mice, caveolin-1 expression is down-regulated while the expression of collagen, tenascin-C, and ASMA and the activation of MEK, ERK, JNK, and Akt are up-regulated. These observations validate the relevance of both SLF as an in vitro model and bleomycin-treated mice as an in vivo model for the fibrosis observed in scleroderma lung disease. When mice received i.p. CSD to reverse the effects of loss of caveolin-1 following bleomycin treatment, the protective effects of CSD observed included inhibition of the increases in the expression of collagen, tenascin-C, and ASMA, and the activation of MEK, ERK, JNK, and Akt; improved tissue morphology; and decreased weight loss. Similar protective effects were observed in bleomycin-treated mice receving intratracheal administration of adenovirus that mediate the expression of caveolin-1 [57].
While CSD clearly seems to act in lung fibroblast cultures and in the bleomycin model by restoring caveolin-1 function and thereby reversing the hyperactivation of a variety of signaling molecules, it may act through a somewhat different mechanism in other disease model systems. In a model system in which colitis is induced using dextran sulfate, caveolin-1 levels increase, the caveolin-1 knockout mouse is resistant to the disease, and CSD provides protection against the disease in wild-type mice [61]. Therefore, in this model CSD acts by inhibiting the function of caveolin-1 rather than by restoring its function. On the one hand, this difference in CSD function may be due to the different cell types being targeted (endothelial cells in colitis; fibroblasts, epithelial cells, and monocytes in lung fibrosis). In addition, just as CSD affected the distribution of caveolin-1 in lung fibroblasts [25], it may act in part by changing the compartmentalization of caveolin-1 in the colitis model.
CAVEOLIN-1 IN LUNG FIBROSIS – INFLAMMATION
While many papers have reported the importance of caveolin-1 in inflammation, most of these papers have focused on caveolin-1 in endothelial cells where it is important in signaling cascades involving mediators such as I-CAM, LPS, and NO [20, 27, 62-66]. Less is known about caveolin-1 in leucocytes. In LPS-stimulated macrophages caveolin-1 inhibits JNK, ERK, and Akt activation, but promotes p38 MAPK activation [57]. In vivo experiments demonstrate roles for caveolin-1 in neutrophil activation, transendothelial migration, and resultant lung inflammation [67]. We observed [25] that the CSD peptide inhibits the accumulation of inflammatory cells in the lung tissue of bleomycin-treated mice, possibly by restoring caveolin-1 function in epithelial cells thereby inhibiting their apoptosis.
Given that lung fibroblasts from human patients and experimental mice with lung fibrosis are deficient in caveolin-1, we examined whether inflammatory cells from subjects with lung fibrosis might also be deficient in caveolin-1. In both scleroderma monocytes and PMNs, caveolin-1 expression was diminished while several members of the MAP kinase family of signaling molecules (ERK, JNK, p38) were hyperactivated. In addition, Cox-2 expression was increased in both monocytes and PMNs and expression of the pro-migratory chemokine receptor CXCR4 and of the inflammatory mediator MMP-9 was increased in scleroderma monocytes [12]. To confirm that the low level of caveolin-1 in scleroderma monocytes is upstream from the other observed alterations in phenotype, cells were treated with CSD which reversed the hyperactivation of ERK, JNK, and p38 and the upregulation of CXCR4 and MMP-9 expression, but did not significantly affect Cox-2 expression [12]. Monocytes from scleroderma patients and from bleomycin-treated mice were similar in that both showed a loss of caveolin-1.
CAVEOLIN-1/CXCL12/ CXCR4 RELATIONSHIP
The peak of collagen deposition during pulmonary fibrosis is preceded by a phase during which inflammatory cells migrate from the peripheral blood into the damaged lung tissue. This migration is mediated by chemoattractant chemokines produced in the lung that bind and activate chemokine receptors on the inflammatory cells. The most effective chemokine for monocytes and fibrocytes is Stromal Cell–Derived Factor-1 (SDF-1, also known as CXCL12) which is produced by many types of stromal cells [68] and binds to chemokine receptor CXCR4. The CXCL12/ CXCR4 axis has been been demonstrated to regulate the migration of monocytes and fibrocytes into the lungs in the bleomycin model [41, 69]. We have validated the importance of this axis in SSc ILD by demonstrating that CXCL12 is expressed at high levels in SSc lung tissue, that CXCR4 is overexpressed on SSc monocytes, and that SSc monocytes are hypermigratory in response to CXCL12 due to their overexpression of CXCR4 [12, 69]. Most important, we have shown that the deficiency in caveolin-1 in SSc monocytes is responsible for their overexpression of CXCR4 and that using CSD to reverse this deficiency reverses the overexpression of CXCR4 and hypermigration exhibited by these cells [69]. We have also used the bleomycin model to demonstrate the importance of this axis in vivo by demonstrating that in bleomycin-treated mice: 1) Circulating monocytes are deficient in caveolin-1 and overexpress CXCR4, 2) CSD reverses the overexpression of CXCR4 by circulating monocytes, and 3) CSD inhibits the accumulation of monocytes and fibrocytes in the lung tissue of bleomycin-treated mice [12, 25, 69].
SSc monocytes also exhibit a different phenotype than normal monocytes, expressing high levels of ColI, CD14, and CD34. This is particularly noteworthy because: 1) It has been proposed that CD14+ monocytes, but not CD14- monocytes, are fibrocyte precursors [14]; and 2) Both CD45+/ColI+/CD14- fibrocytes and CD45+/ColI+/CD14+ cells recently named collagen-producing monocytes [70] are present at higher levels in the blood and lung tissue of SSc patients than in normal samples [69, 70]. The greater fraction of CD14+ cells raises the possibility that the percentage of SSc-ILD monocytes able to differentiate into fibrocytes is higher than the percentage for normal monocytes. The greater fraction of CD45+/ColI+/CD14- and CD45+/ColI+/CD14+ cells suggests that SSc-ILD monocyte preparations contain a significant number of cells that are already partially or fully differentiated into fibrocytes and collagen-producing monocytes. In summary, these results suggest that low caveolin-1 in SSc monocytes contributes to ILD via effects on cell migration and phenotype, and that the hyperaccumulation of fibrocytes and collagen-producing monocytes in SSc-ILD lung tissue may result from the altered phenotype and migratory activity of these cells and/or their monocyte precursors.
We propose that the regulation of CXCR4 expression by caveolin-1 is important in the trafficking of fibrocytes and/or their precursors from the bone marrow into the circulation as well as from the circulation to damaged lung tissue as described above. A cartoon summarizing our hypothesis is presented in Fig. (2).
CAVEOLIN-1 AND TGF-β SIGNALING
TGF-β signaling is extremely well studied, especially in the regulation of collagen expression [71]. There are multiple points of intersection between TGF-β and caveolin-1 signaling. First of all, TGF-β inhibits caveolin-1 expression in a variety of cell types including fibroblasts (both lung and dermal) and monocytes [12, 57, 72]. These observations suggest that the TGF-β-rich milieu in the blood and tissues of scleroderma patients leads to the reduction of caveolin-1 expression in fibroblasts and monocytes and thereby to the altered behavior of these cells that promotes fibrosis. Del Galdo et al. also showed that caveolin-1 modulates TGF-β signaling in dermal fibroblasts by inhibiting Smad3 phosphorylation and its translocation to the nucleus [72].
Caveolin-1 also regulates TGF-β signaling via its effects on the endocytosis of TGF-β ligand-receptor complexes. TGF-β receptors are present in both caveolin-1-rich lipid rafts and early endosomes [73, 74]. The internalization of TGF-β receptor is dependent on the function of caveolin-1 in lipid rafts and clathrin in early endosomes. Early endosomal internalization increases TGF-β signaling while caveolin-1-dependent internalization in lipid rafts leads to receptor degradation, thereby inhibiting TGF-β signaling [73]. TGF-β and other growth factors and ECM molecules promote TGF-β signaling by shifting TGF-β receptors from the inhibitory caveolar lipid raft fraction to the activating early endosomal fraction [73, 75, 76]. Therefore, it is likely that the loss of caveolin-1 that occurs in fibroblasts in fibrotic tissues will promote TGF-β signaling by inhibiting TGF-β receptor turnover in lipid rafts and by promoting the localization of TGF-β receptors to the activating environment in early endosomes.
CAVEOLIN-1 IN DERMAL FIBROSIS
Some studies show a similar role for caveolin-1 in dermal fibrosis to that described above for lung fibrosis, while other studies show significant differences between dermal and lung fibrosis. On the one hand, it has been reported that dermal fibroblasts, like lung fibroblasts, from SSc patients and caveolin-1 KO mice are deficient in caveolin-1 and overexpress collagen [77]. Both the basal and the TGF-β-induced overexpression of collagen by these cells can be inhibited using CSD. Similarly, dermal fibroblasts from keloids are deficient in caveolin-1 and overexpress collagen in response to TGF-β via a MAP kinase signaling cascade that can be inhibited with CSD [78]. In addition to being a negative regulator of collagen expression in dermal fibroblasts, caveolin-1 is also a negative regulator of the collagenase MMP-1 [79].
In contrast to studies on lung fibroblasts, other studies found higher caveolin-1 levels in scleroderma dermal fibroblasts than in normal dermal fibroblasts [5] and that in normal dermal fibroblasts caveolin-1 overproduction enhanced collagen expression while caveolin-1 depletion inhibited collagen expression [80]. Despite the disparity in caveolin-1 levels, enhanced MEK and ERK activation and enhanced collagen expression that could be blocked with MEK inhibitor U0126 were observed both in scleroderma and normal lung and dermal fibroblasts [5]. Therefore, the results are still consistent with the idea that in both lung and dermal fibroblasts, decreasing caveolin-1 expression increases MAP kinase activation and increasing MAP kinase activation increases collagen expression. The distinction is that the baseline level for caveolin-1 in normal dermal fibroblasts is higher than in normal lung fibroblasts and even higher in scleroderma dermal fibroblasts [5] (possibly due to the high level of expression of PKCα in these cells). The association of high caveolin-1 levels and high collagen expression in scleroderma dermal fibroblasts may be related to the observation that in dermal fibroblasts caveolin-1 overexpression activates Akt and thereby enhances collagen expression [80]. Alternatively, another upstream regulator of MAP kinase activation may be dominant in dermal fibroblasts, while caveolin-1 is dominant in lung fibroblasts. Whether these differences between lung and dermal fibroblasts exist in vivo or are generated during the isolation and culture of the cells is an important question that will require further study.
CAVEOLIN-1 AS A TARGET FOR DISEASE TREATMENTS
The use of caveolin-1 as a molecular therapeutic target for treating fibrosis may be a good idea because caveolin-1 is a master regulator of several signaling pathways. The pharmaceutical industry has spent billions of dollars to find strong, specific inhibitors of individual signaling pathways and they have succeeded in designing these inhibitors. Yet they have had little or no success in developing a treatment for fibrosis and only limited success in treating cancer. These poor outcomes may result from a flaw in the paradigm, i.e. it may not be a good idea to treat certain diseases with a strong, specific inhibitor of a single signaling pathway. The use of targets such as caveolin-1 that may be moderate inhibitors of multiple, parallel pathways may not be as elegant as the use of a strong, specific inhibitor of a single signaling pathway. However, the functional up-regulation of a broad-spectrum endogenous inhibitor of several pathways such as caveolin-1 may be a more effective way to treat certain diseases, particularly fibrosis.
ACKNOWLEDGEMENTS
Our work on caveolin-1 in lung fibrosis has been supported by: Grants to ET from the Scleroderma Foundation and the NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases (R03 AR056767 and K01 AR054143) and to SH from the National Heart, Lung, and Blood Institute (R01 HL73718), the National Center for Research Resources (UL1 RR029882), and the Department of Defense.
CONFLICT OF INTEREST
Declared none.

