All published articles of this journal are available on ScienceDirect.

Sphingolipid Regulation of Tissue Fibrosis
Abstract
Bioactive sphingolipids, such as sphingosine 1-phosphate (S1P), dihydrosphingosine 1-phosphate (dhS1P) and ceramide, regulate a diverse array of cellular processes. Many of these processes are important components of wound-healing responses to tissue injury, including cellular apoptosis, vascular leak, fibroblast migration, and TGF-β signaling. Since over-exuberant or aberrant wound-healing responses to repetitive injury have been implicated in the pathogenesis of tissue fibrosis, these signaling sphingolipids have the potential to influence the development and progression of fibrotic diseases. Here we review accumulating in vitro and in vivo data indicating that these lipid mediators can in fact influence fibrogenesis in numerous organ systems, including the lungs, skin, liver, heart, and eye. Targeting these lipids, their receptors, or the enzymes involved in their metabolism consequently now appears to hold great promise for the development of novel therapies for fibrotic diseases.
INTRODUCTION
The response to tissue injury involves a complex series of biological responses which, if appropriate in timing, magnitude and balance, restore normal tissue structure and function. When dysregulated or over-exuberant, however, these responses can result in fibrosis, characterized by the pathological accumulation of fibroblasts and myofibroblasts, and the extracellular matrix that they synthesize. Fibrosis is frequently observed following injury to the lungs, skin, liver, heart, and eye. In many fibrotic diseases, such as idiopathic pulmonary fibrosis (IPF) and systemic sclerosis (SSc), the replacement of normal tissue with fibrosis is progressive and unrelenting, eventually leading to end-stage organ dysfunction and death [1,2]. Since tissue fibrosis may be difficult to reverse once established, strategies aimed at preventing fibrosis by interrupting the upstream biological processes that contribute to fibrogenesis are likely to have the greatest therapeutic impact.
Sphingolipids were originally identified as structural components of biological membranes. Many of them, most notably sphingosine 1-phosphate (S1P) and ceramide, are also now recognized to be important mediators of many basic cellular processes involved in tissue responses to injury, such as cell migration, survival, contraction, proliferation, gene expression, and cell-cell interactions [3-5]. By virtue of their ability to regulate these processes, there has been substantial recent interest in the ability of sphingolipids, particularly S1P, to regulate tissue fibrosis in various organ systems. S1P exerts its cellular effects predominantly through interactions with a family of specific cell surface G protein-coupled receptors, labeled S1P1-5 [3,6]. Especially relevant to wound healing and fibrotic responses to tissue injury, S1P signaling through its receptors has been shown to regulate epithelial cell apoptosis, fibroblast migration and myofibroblast differentiation, vascular permeability, and TGF-β signaling in vitro [7-12]. Furthermore, numerous recent studies have suggested important roles for S1P in the development of fibrosis in the lungs, skin, liver, heart and eye, and alterations in circulating S1P levels have been observed in various human diseases characterized by tissue fibrosis [13-19]. In this review we discuss the current literature implicating sphingolipid signaling in the regulation of fibrogenesis in multiple organs (Table 1). We focus predominantly on S1P, but discuss evidence that ceramide and dihydrosphingosine 1-phosphate (dhS1P) regulate fibrogenesis as well.
Organ Fibrosis in which S1P, dhS1P, Ceramide and/or their Synthetic Enzymes have been Implicated
| Affected Organ | Sphingolipid/ Enzyme Implicated | Receptor(s) Implicated | Pro- or Anti-Fibrotic Role | Proposed Mechanism(s) | References |
|---|---|---|---|---|---|
|
|
|||||
| Lung fibrosis | S1P | S1P1 | Anti-fibrotic | Attenuation of vascular leak and intra-alveolar coagulation | [13] |
| Acid sphingomyelinase (ASM) | ? | Pro-fibrotic | Promotion of cellular apoptosis and fibroblast collagen synthesis | [76] | |
|
|
|||||
| Skin fibrosis/ Scleroderma | S1P | S1P1,2 | Pro-fibrotic | Cross-activation of fibroblast TGF-β/ Smad signaling; myofibroblast differentiation | [78-81] |
| dhS1P | S1P1,2 | Anti-fibrotic | Inhibition of fibroblast TGF-β/Smad signaling through PTEN upregulation | [14, 81] | |
|
|
|||||
| Liver fibrosis | S1P | S1P2,3 | Pro-fibrotic | Hepatic stellate cell accumulation and activation; homing of bone marrow-derived cells to the injured liver | [15, 84-88] |
| Ceramide/Acid sphingomyelinase (ASM) | ? | Pro-fibrotic | Inhibition of fibroblast TGF-β/Smad signaling through PTEN upregulation | [89-92] | |
|
|
|||||
| Cardiac fibrosis | S1P | S1P2,3 | Pro-fibrotic | Promotion of hepatocyte apoptosis; hepatic stellate cell proliferation and activation | [16, 75] |
|
|
|||||
| Retinal fibrosis | S1P | ? | Pro-fibrotic | Promotion of retinal pigmented epithelial cell proliferation, myofibroblast differentiation, and collagen synthesis | [17, 93] |
SPHINGOLIPID METABOLISM
Sphingolipids are a class of complex, structurally-related compounds derived from sphingoid bases, with hundreds of known class members. The first sphingoid base characterized, and the most common in mammalian organisms, is sphingosine [20,21]. The enzymatically catalyzed pathways through which sphingosine and many of the most commonly studied signaling sphingolipids, including S1P, dhSIP, and ceramide, are converted into each other are depicted in Fig. (1). Given the pleiotropic effects of these sphingolipids on physiological and pathological cellular functions, altering their levels by targeting the enzymes involved in their metabolism holds great therapeutic potential for human diseases.
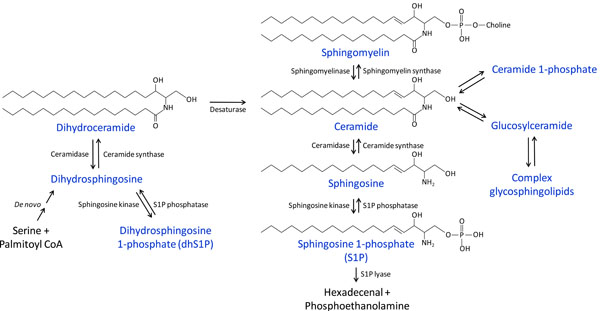
Overview of sphingolipid metabolism.
Ceramide is at the center of these metabolic pathways (Fig. 1). It can be generated de novo from serine and palmitoyl-CoA through several intermediate steps, the last of which is the desaturation of dihydroceramide. Ceramide alternatively can be formed from the catabolism of sphingomyelin by sphingomyelinases (SMase) or the breakdown of complex glycosphingolipids. Conversely, ceramide can be converted back to sphingomyelin by the addition of a phosphocholine group by sphingomyelin synthases. Ceramide can be phosphorylated to ceramide 1-phosphate by ceramide kinase, or it can be glycosylated by glucosyl- or galactosylceramide synthases. Ceramide can also be metabolized by ceramidases to form sphingosine, which can in turn be phosphorylated by sphingosine kinases (Sphk), producing S1P. S1P can either be converted back to sphingosine by the action of S1P phosphatases or lipid phosphate phosphatases (LPPs) [22], or it can be degraded by S1P lyase. Similar enzymatic pathways catalyze the interconversion of dihydroceramide, dihdydrosphingosine, and dihydrosphingosine 1-phosphate (dhS1P), as also shown in Fig. (1).
SPHINGOLIPIDS AND LUNG FIBROSIS
Devastating fibrotic lung diseases such as IPF may result from over-exuberant wound-healing responses to common insults to the lung, such as from inhaled particulates, viral infections, or aspiration of refluxed gastric acid [23-25]. Biological responses to injury which, if dysregulated, may contribute to the development of these diseases include: 1) epithelial cell death/apoptosis, 2) increased vascular permeability, 3) inflammatory cell recruitment, 4) extravascular coagulation, 5) activation of transforming growth factor-β (TGF-β) and other pro-fibrotic mediators, 6) fibroblast recruitment, proliferation, persistence and activation to myofibroblasts, and 7) synthesis of collagen and other extracellular matrix proteins [26-29]. Better understanding of the molecular mediators and pathways that regulate these aberrant wound-healing responses should therefore lead to novel therapeutic targets for fibrotic diseases. In support of this approach, multiple investigations in animal models of lung fibrosis have demonstrated that targeting one or more of these wound-healing responses can prevent the development of fibrosis, and in some cases reverse established fibrosis [30-36].
Recent data indicates that S1P, signaling through the S1P1 receptor, plays a key role in regulating the development of fibrosis after lung injury. We demonstrated that inhibition of S1P-S1P1 signaling resulted in increased pulmonary fibrosis in the bleomycin mouse model of this disease, indicating that this pathway functions in the lung as an endogenous inhibitor of fibrogenesis [13]. While there are numerous potential mechanisms to explain the anti-fibrotic effect of S1P-S1P1 signaling, we hypothesize that it is largely due to the ability of this pathway to attenuate vascular leak after lung injury. S1P has been established as a key regulator of vascular permeability, and it limits the vascular leak characteristic of the early, exudative response to lung injury. S1P’s ability to regulate vascular permeability in the lung appears to be attributable to signaling specifically through S1P1. Activation of S1P1 on endothelial cells by S1P or S1P1 agonists induces cytoskeletal rearrangements which promote formation of intercellular adherens and tight junctions, resulting in enhancement of endothelial barrier function [8,37-41]. Mice which are genetically deficient for the S1P-producing enzyme sphingosine kinase 1 (Sphk1) develop increased vascular leak after lung injury [42,43]. Genetic deletion of both sphingosine kinases (Sphk1 and Sphk2) results in a complete lack of circulating S1P and increased vascular leak in multiple organ systems, even in the absence of tissue injury [44]. Interrupting S1P-S1P1 signaling with receptor antagonists, or with S1P analogs that act as functional antagonists of S1P1 with sustained administration, similarly increases vascular leak [13,45]. Conversely, short-term treatment with S1P or S1P1 receptor agonists has been shown to prevent vascular leak in numerous animal models of acute lung injury, as well as models of renal and cutaneous injury [39,41,45-51].
Vascular leak is one of the hallmarks of tissue injury, and it contributes significantly to organ dysfunction [52,53]. In the lung, persistent vascular leak may also contribute to the subsequent development of lung fibrosis. Increased lung capillary-alveolar permeability has been demonstrated to be present in the lungs of IPF patients and to predict worse outcomes [54,55]. One consequence of increased vascular permeability in the lungs is extravasation of clotting factors into the airspaces, where exposure to tissue factor in injured alveoli can initiate the coagulation cascade. There are extensive data indicating that activation of the coagulation cascade is a critical component of the fibrotic response to lung injury [30,32,33,56-63]. The resulting deposition of fibrin is thought to provide a provisional matrix through which fibroblasts migrate during tissue repair [52]. Fibrin in the airspaces may also promote epithelial-to-mesenchymal transition, further contributing to fibroblast accumulation [64]. If excessive or excessively persistent following injury, fibrin deposition consequently may contribute to the development of fibrosis [65]. Fibrin deposition is not absolutely required for the development of fibrosis [59,66], however, and extravascular activation of the coagulation cascade may contribute to fibrogenesis through other mechanisms. In addition to their role in the generation of fibrin, thrombin and other coagulation cascade proteinases also activate a family of cell-surface receptors, the proteinase-activated receptors (PARs). Activation of these receptors may promote fibrosis independently of fibrin generation, through the induction of mediators such as PDGF, CTGF and MCP-1/CCL2 or through the activation of latent TGF-β [33,63,67]. S1P-S1P1signaling may therefore attenuate the development of fibrosis after lung injury by limiting vascular leak and the accompanying extravasation of coagulation proteinases into the injured airspaces.
In vitro studies have shown that S1P is capable of mediating other processes which may contribute to the development of lung fibrosis, although some of these studies have produced conflicting data. For example, S1P has been shown to both potentiate and inhibit fibroblast chemotaxis [9,68-70]. S1P has also been shown to have cell-specific effects on apoptosis, either inducing apoptosis or promoting survival in different cell types [11,12,71,72]. S1P could therefore potentially contribute to the increased epithelial cell apoptosis and decreased fibroblast apoptosis observed in lung fibrosis [73]. Lastly, S1P has been shown to play a role in TGF-β-induced differentiation of fibroblasts into myofibroblasts in vitro, another process critically involved in fibrogenesis [10,74,75].
Further evidence that sphingolipids may regulate the development of lung fibrosis comes from recent findings that activity of the sphingomyelin-degrading enzyme acid sphingomyelinase (ASM) is increased in the lungs of mice after intratracheal bleomycin challenge, and that mice genetically deficient for this enzyme exhibit attenuated lung fibrosis in this model [76]. Interestingly, despite the increased lung ASM activity seen in wild-type mice, lung ceramide levels actually decreased after bleomycin challenge, which may have been due to the simultaneous upregulation of the ceramide-degrading enzyme, acid ceramidase [76].
SPHINGOLIPIDS AND SYSTEMIC SCLEROSIS
Sphingolipids have also been implicated in the pathogenesis of systemic sclerosis (SSc). SSc, or scleroderma, is an autoimmune connective tissue disease characterized by widespread vasculopathy and tissue fibrosis [2]. Although the hallmark of this disease is progressive thickening and fibrosis of the skin, fibrosis in SSc can involve many different organ systems, including the lungs, heart, kidneys, and digestive tract [2]. Like IPF, SSc is a progressive and often fatal disorder. Although there is an inflammatory component to the pathological changes seen in SSc, again similar to IPF it is largely refractory to conventional anti-inflammatory and immunosuppressive medications [2]. TGF-β and its signaling pathways are thought to play a central role in the fibrogenesis seen in SSc [77]. Numerous studies have shown that sphingolipids, most notably S1P and dhS1P, can regulate TGF-β signaling pathways in skin cells in vitro, and there are recent data implicating altered sphingolipid signaling in the pathogenesis of human SSc.
In contrast to the role that S1P appears to play limiting the development of pulmonary fibrosis, S1P may promote the development of dermal fibrosis in SSc. S1P has been shown to induce activation of the canonical TGF-β signaling pathway in numerous cell types, including skin fibroblasts [78-81]. Specifically, S1P has been shown to induce phosphorylation of Smad2 and Smad3 and upregulate expression of Smad-dependent genes. This process appears to be independent of TGF-β itself, but dependent on the S1P receptors, suggesting that there is crosstalk between the S1P and TGF-β signaling pathways. S1P1, S1P2 and S1P3 have all been implicated in TGF-β signaling pathway activation by S1P, and the specific receptor(s) involved are likely cell type-dependent. Further support for S1P playing a pro-fibrotic role in SSc comes from recent data showing that S1P levels are increased in the serum of SSc patients compared with matched controls [18]. Moreover, since S1P is known to regulate immunity and endothelial cell function, it may also play central roles in the autoimmune and vascular manifestations of SSc [82].
Dihydrosphingosine 1-phosphate (dhS1P), which is also produced by sphigonsine kinase and binds to the S1P receptors (Fig. 1), has been shown to have opposing effects to those of S1P on TGF-β signaling in skin fibroblasts, and may therefore limit the development of dermal fibrosis [81]. dhS1P inhibits TGF-β-mediated Smad2 and 3 phosphorylat-ion and collagen synthesis in a manner dependent on PTEN, a tumor suppressor gene which has anti-fibrotic properties [83]. dhS1P promotes formation of a PTEN-Smad2/3 complex, which results in the dephosphorylation of Smad2 and Smad3 and reduced expression of Smad-dependent genes [81]. Reduced PTEN expression has been found in skin fibroblasts isolated from SSc patients, as well as in myofibroblasts in lung sections from IPF patients [14,83]. Whereas treatment of SSc fibroblasts with S1P resulted in further reduction of PTEN levels and increased collagen I production, treatment with dhS1P restored PTEN levels to normal and inhibited collagen synthesis [14].
SPHINGOLIPIDS AND FIBROSIS IN OTHER ORGAN SYSTEMS
The critical role that sphingolipids play in regulating tissue fibrosis does not appear to be limited to the lung and skin, as these signaling lipids have been implicated in liver, cardiac, and retinal fibrosis as well. S1P has been shown to be pro-fibrotic in two different mouse models of liver fibrosis. In carbon tetrachloride (CCl4)-induced liver fibrosis, S1P signaling through S1P2 appeared to promote fibrosis by mediating the accumulation of matrix-producing hepatic stellate cells and/or myofibroblasts [84]. In cholestasis-induced liver fibrosis, S1P levels and S1P3 receptor expression were found to be elevated in the liver, and pharmacological inhibition of S1P3 attenuated the development of fibrosis in this model [15]. Attenuation of fibrosis by S1P3 inhibition in the cholestasis model was attributed to decreased S1P3-dependent homing of pro-fibrotic, bone marrow-derived stem cells to the liver after injury. S1P has also been shown to have effects on hepatic stellate cell and myofibroblast survival, proliferation, migration and activation in vitro [85-88]. Lastly, alterations in circulating and liver S1P levels and S1P receptor expression patterns have been found in humans with liver fibrosis [19,87].
Ceramide has also been investigated as a potential regulator of liver fibrosis. Ceramide has been shown to mediate hepatocellular apoptosis [89,90], and inhibition of acid sphingomyelinase (ASM) has been shown to reduce liver ceramide levels and attenuate liver fibrosis in a rat model of Wilson’s disease [91]. ASM haploinsufficient mice have also been shown to be protected from the development of liver fibrosis in both cholestasis- and CCl4-induced models of this disease [92]. Lastly, ASM activity has been found to be elevated in the blood of humans with Wilson’s disease, and ASM expression has been found to be elevated in the livers of patients with nonalcoholic steatohepatitis (NASH) [91,92].
In addition to its pro-fibrotic effects noted in the liver, S1P-S1P3 signaling has also been found to promote fibrosis in the heart. Mice which overexpress the S1P-producing enzyme, sphingosine kinase 1 (Sphk1), develop spontaneous cardiac fibrosis. The ability of Sphk1 overexpression to produce cardiac fibrosis was attenuated when these mice were crossed to S1P3-deficient mice, suggesting that S1P’s pro-fibrotic effects in the heart are mediated at least in part by S1P3 [16]. S1P-S1P2 signaling has also been shown to induce myofibroblast differentiation and increased collagen production in rat cardiac fibroblasts [75].
Finally, pro-fibrotic activity of S1P has also been observed in the eye. S1P stimulates retinal pigmented epithelial (RPE) cell proliferation, myofibroblast differentiation, and collagen synthesis, and anti-S1P neutralizing antibodies were shown to protect against the development of subretinal fibrosis [17,93].
CONCLUSIONS
Sphingolipids, such as S1P, dhS1P, and ceramide play key roles in regulating numerous cellular processes, many of which have been implicated both in physiological wound-healing responses and the pathological wound-healing responses thought to contribute to multiple fibrotic diseases. As discussed above, there are accumulating data that sphingolipids play important roles in regulating the development of tissue fibrosis in numerous organ systems, including the lungs, skin, liver, heart, and eye (Table 1). In the lung, S1P signaling through S1P1 appears to protect against the development of fibrosis, likely through to its ability to limit vascular leak during the early, exudative response to lung injury. Conversely, S1P appears to promote fibrosis in other organ systems, likely through activation of TGF-β signaling pathways and/or by promoting fibroblast migration. Interestingly, dhS1P, which also signals through the S1P receptors, appears to oppose the pro-fibrotic effects of S1P in the skin. Lastly, data obtained from pharmacologic or genetic manipulation of acid sphingomyelinase (ASM) suggest that ceramide may also regulate fibrosis in the lungs and liver. Although further study is needed to better elucidate the mechanisms and specific pathways involved in the pro- and anti-fibrotic effects of these and other related sphingolipids, it is becoming increasingly clear that targeting these lipids, their receptors, or the enzymes involved in their metabolism holds great promise for the development of much-needed therapies for fibrotic diseases.
ACKNOWLEDGEMENT
This work was supported by NIH K08 HL105656 to B.S.S, and NIH R01 HL108975, NIH R01 HL095732, a Scleroderma Research Foundation Grant, and a Nirenberg Center for Advanced Lung Disease Grant to A.M.T.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.

