All published articles of this journal are available on ScienceDirect.

LETTER TO THE EDITOR Atypical Granulomatous Myositis and Pulmonary Sarcoidosis
Authors Info & Affiliations
SIR,
Up to 80% of patients with sarcoidosis show muscular involvement, but symptomatic myopathy (sarcoid myopathy, SM) is rare [1, 2]. We herein report an interesting case of a patient with long history of muscle weakness diagnosed with SM who presented pulmonary sarcoidosis at a later stage.
A 45-year-old Brazilian white female was admitted with a four-year history of progressive proximal muscle weakness and myalgia. A chest radiogram was normal at the beginning of the clinical presentation. A few weeks before hospital admission, she developed diffuse weakness, dysphagia and exertion dyspnea. A physical examination showed weakness of facial and proximal muscles and severe wasting of hand muscles. Her family history was negative for neuromuscular diseases. The erythrocyte sedimentation rate was 92 mm in the first hour, and the serum creatine kinase level was high (6,683 IU/L, with a normal range being up to 176 IU/L). Antinuclear antibodies (1/640, granular pattern) and circulating anti-Ro/SSA antibodies (56.7 IU) were present. No other autoantibodies were found. A dried blood spot test for Pompe disease was negative. Neurophysiology assessment showed myopathic changes. A muscle biopsy performed 4 years after disease onset showed increased variation in fibre size, increased connective tissue, internal nuclei, occasional atrophic fibres and necrotic fibres. There was a prominent endomysial and perimysial inflammatory infiltrate composed of lymphoctes and macrophages with the formation of non-necrotic granulomas including small numbers of multinucleate giant cells (Fig. 1). A second piece of tissue taken from the same muscle showed no inflammatory features. Investigations for tuberculosis, fungi and brucellosis were negative. Lung computed tomography performed after 4 years of disease onset showed typical bilateral hilar lymphadenopathy and interstitial pneumonitis (“ground-glass” aspect in lower lobes) compatible with sarcoidosis. A restrictive pattern was seen on spirometry. Following the diagnosis, she received high doses of steroids. She has received IV immunoglobulin, and immunosuppressant (IV cyclophosphamide pulse therapy) since the beginning of the clinical presentation with no clinical response. Unfortunately, the patient died from bronchopneumonia approximately 5 years after initial presentation.
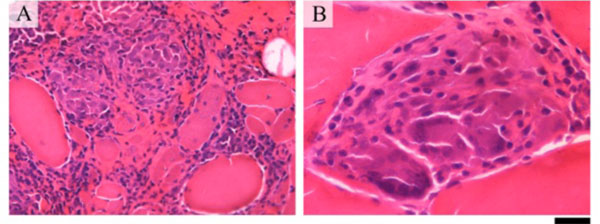
Muscle biopsy showing inflammatory infiltration of the perimysium and endomysium including non-necrotic granulomas (A). Multinucleate giants cells are seen within the granuloma (B). The bar represents 50 μm in A; and 20 μm in B.
The described patient presented muscle weakness as the main manifestation of systemic sarcoidosis. An early muscle biopsy is highly recommended to diagnose SM. Three different forms of SM are described: acute myositis, palpable muscle nodules and chronic myopathy. Our patient presented the latter, which is the most common [1, 2]. The cause of her muscle symptoms was probably SM/granulomatous myositis (GM), which later progressed to pulmonary sarcoidosis refractory to immunosuppressive therapy. ANA and anti-Ro/SSA were detected by the time the systemic disease was evident, but the patient did not show mucosal features typical of Sjögren syndrome. Anti-Ro/SSA antibodies have indeed been rarely reported in patients with sarcoidosis in the absence of Sjögren syndrome [3]. As opposed to descriptions in pulmonary sarcoidosis, whereby a Th1 cytokine response is characteristic, a dominant Th2 pattern is reported in patients with SM [4].
GM is a rare disorder, which can be idiopathic, but more frequently is related to sarcoidosis [5]. The clinical features of GM are non-specific including myalgia and weakness presenting a diagnostic challenge. Distal muscle involvement in GM may simulate inclusion body myositis [6]. GM can also mimic slowly progressive lower motor neuron disease [7] and polymyalgia rheumatic [8], or be associated with myasthenia gravis [9] and mixed connective tissue disease [10]. GM has also been reported in patients with graft-versus-host disease [11], Crohn disease [12], primary biliary cirrhosis, pancytopenia, and thymoma [13]. Of note, GM was the first manifestation of an aggressive CD30+ extra-nodal NK/T-cell lymphoma [14]. Infection diseases should be excluded including brucellosis [15], Pneumocystis carinii [16], and tuberculosis [17].
Four out of 10 GM patients with myopathy reported in 1998 had systemic sarcoidosis [18]. Such patients had severe proximal weakness and were steroid-responsive as opposed to patients with idiopathic GM who had mostly distal involvement and were refractory to steroids [18]. A second GM series of symptomatic cases was published in 2007 [19]. The authors described 8 SM cases [19]. Distal involvement was also rare among the SM cases [19]. Our patient showed an atypical form of SM, considering the presence of generalized muscle weakness with facial and distal muscle involvement, pulmonary sarcoidosis and anti-Ro/SSA antibodies.
A number of pathological findings have been reported in GM and SM including granulomatous endomysial and perimysial inflammation without necrosis. Typical inflammatory cells present are macrophages and T4 lymphocytes surrounded by T8 lymphocytes in mature lesions [18, 20]. Necrotic and regenerating fibres may be seen. The pathological findings can be patchy and therefore may not be observed in the biopsy [18]. Sarcolemmal MHC class I up-regulation has been variably reported [4, 20]. Chronic disease may progress to muscle fibrosis [4]. Muscle magnetic resonance imaging and ultrasound may help in the selection of biopsy site, differentiation of SM types and monitoring of therapy [21]. Whole body fluorodeoxyglucose positron emission tomography can also be useful to direct biopsies in suspected sarcoidosis by revealing areas of increased metabolism [22].
In conclusion, the reported case suggests that SM/GM may precede systemic sarcoidosis. Presentation may be atypical as in our patient, who showed facial and distal muscle involvement and anti-Ro/SSA antibodies.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

