All published articles of this journal are available on ScienceDirect.

Allele Specific Expression of MICA Variants in Human Fibroblasts Suggests a Pathogenic Mechanism
Authors Info & Affiliations
Abstract
The major histocompatibility complex class I chain-related gene A (MICA) is involved in immune responses of both nature killer (NK) cells and subsets of T cells with its receptor NKG2D. MICA is highly polymorphic in sequence which leads to MICA protein variants with distinct features. Specific polymorphisms of MICA have been associated with inflammatory diseases, including ankylosing spondylitis (AS), ulcerative colitis (UC) and Behçet’s disease. Studies herein characterize expression features of three MICA variants including MICA*008, a common variant in general population, and *MICA*007 and *019, which are associated with susceptibility to inflammatory diseases. MICA*019 was highly expressed on the surface of fibroblasts whereas expression of MICA*007 was the lowest in the culture supernatant. MICA*008 had low cell surface expression but was the only MICA allele in which exosomal material was detected. Surface or membrane-bound MICA activates NKG2D-mediated cytotoxicity, whereas soluble and exosomal MICAs down-regulate NKG2D. Therefore, comparisons of these three MICA variants in fibroblasts provides insight into understanding how MICA associated immune responses could be regulated to influence levels of inflammation.
INTRODUCTION
The major histocompatibility complex class I chain-related gene A (MICA) is highly polymorphic. MICA with more than 100 identified alleles [http://www.ebi.ac.uk/ipd/ imgt/hla/]. Although, the functional significance of these alleles has not been fully defined, some have been associated with chronic inflammatory diseases. In particular, MICA*007 and *019 alleles were associated with ankylosing spondylitis (AS) [1], MICA*007 also was associated with ulcerative colitis (UC) [2], and MICA*019 with Behçet’s disease [3]. This association indicates that these two MICA alleles may be important in regulation of inflammation. Compared to the most common MICA*008 allele, several nucleotides differ in the MICA*007 and *019 alleles with the most prominent sequence variation at codon 293-297, encoding four (MICA-A4) or five (MICA-A5) alanine residues in the transmembrane domain of MICA, respectively. By contrast, the MICA*008 sequence contains an insertion of guanine at codon 295 that results in a premature stop codon at position 304, which in turn encodes a truncated MICA protein lacking part of the transmembrane domain and the whole cytoplasmic tail [4].
MICA protein is expressed as a membrane-bound protein in selective cells such as gut epithelial and fibroblasts. Under cellular stress conditions, such as infections, tissue injury, pro-inflammatory signals, and malignant transformation [5-1], MICA interacts with its receptor NKG2D found on natural killer (NK) cells, NK T cells, γδ T cells, αβ CD8+ T cells, and a minor immune-regulatory subset of CD4+ T cells [10-18]. Binding of MICA (membrane-bound MICA) with NKG2D triggers cell-mediated cytotoxicity and cytokine release from NK cells and T cells [9, 10, 19, 20]. On the other hand, the proteolytic cleavage of MICA proteins from expressing cells, termed as MICA shedding produces soluble MICA that may control the immune process by down-modulating NKG2D expression [21, 22], and facilitate expansion of an immunosuppressive CD4+ T-cell subset [15]. In addition, MICA can be excreted in exosomes which can also down-regulate NKG2D activity [23]. It was reported that MICA*008 protein was preferentially released from cells in exosomal form [23]. Therefore, the balance between membrane-bound MICA and soluble MICA/exosomal MICA may control the outcome of immune function via NKG2D regulation. Studies of MICA*007 and *019 protein variants in comparison with the most common allele, MICA*008 in expression and secretion in human fibroblasts may help understand potential functional significance of disease-associated MICA alleles.
MATERIALS AND METHODS
MICA Clones
The full length of MICA*007:01 was synthesized and inserted into pUC57 by GenScript (Piscataway, NJ). The full length of MICA*008:01 and 019:01 in pCDNA3.1 were gifts from Dr. Mar Vales-Gomez, Department of Immunology and Oncology Centro Nacional De Biotecnologia (CNB) Spain. All of three MICAs were subcloned into pEGFPC3 to form pEGFPC3-MICA clones by Xho I and BamH I sites. All clones of pEGFPC3-MICAs were confirmed by Sanger sequencing.
Fibroblast Cultures and Transfection
Primary human fibroblasts were grown in Hyclone DMEM/high glucose culture medium (GE Healthcare Life Sciences) plus 10% FBS (Gemini) and 1x penicillin-strep (Life Technologies) until 70-80% confluence. For transfection of each MICA clone, a total of 5x105 fibroblasts were suspended in 82ul neucleofector and 18ul supplement using nucleofector kit (Lonza, amaxa), and then mixed with 5ug of the pEGFPC3, pEGFPC3-MICA*007, pEGFPC3-MICA*008 and pEGFPC3-MICA*019 respectively. Amaxa biosystem Nucleofector II (program U23) was used for electroporation. The transfected fibroblasts were split into two small culture dishes (60x15mm) with 2 ml culture medium without any antibiotics, and were grown under standard culture conditions at 37°C in a humid atmosphere containing 5% CO2. The culture medium was changed after 24-hours transfection. The fibroblasts were harvested for the further analysis after 48-hours transfection. We also performed antibiotic screen for neomycin resistance as a dominant selectable marker for selection on the pEGFPC3. Amazingly, the results were very consistent.
Flow Cytometry Studies for Membrane-Bound MICA
Fibroblasts were rinsed with PBS and detached with Accutase solution (Sigma). Cells were then washed with PBS/2%FBS, and stained for either surface or intracellular MICA in conjunction with GFP expression. For surface staining, cells were incubated with MICA-APC monoclonal antibodies (Biolegend) for 30mins at 4°C, washed with PBS/2%FBS. For intracellular staining, cells were first fixed with Cytofix/Cytoperm solution (BD Biosciences) for 30mins at 4°C then washed with Perm/Wash buffer. Cells were then incubated with MICA-APC monoclonal antibodies for 30mins at 4°C then washed with Perm/Wash buffer. Data were acquired with Gallios Flow Cytometer and analyzed with Kaluza1.2 (Beckman-Coulter). For background subtraction, we examined two controls including the fibroblasts without transfection and transfected with pEGFPC3 only. Assays were done in triplicate.
ELISA Measurement for Soluble MICA
Each 0.4 ml of culture medium from cultured the fibroblasts alone and the fibroblasts transfected with pEGFPC3, pEGFPC3-MICA*007, pEGFPC3-MICA*008 and pEGFPC3-MICA*019 were collected, respectively. The supernatants were concentrated into 0.2 ml by Amicon@Ultra (Millipore UFC500324, 3K membrane) according to the manufactory instruction. The concentration of soluble MICAs with duplication were determined by the human MICA ELISA kit (RayBio, Catalog: ELH-MICA).
Exosomal MICA
A total of 4x106 fibroblasts were transfected with each MICA construct in pCDNA3.1. The media from the cultured fibroblasts were used to extract whole exosomes. Briefly, culture medium was centrifuged at 5,000 x g for 10 minutes to remove cell debris. The supernatant was mixed with the exosome isolation reagent (Life Technologies, Catalog: 4478359) and incubated overnight in 4°C. The samples were centrifuged at 10,000 x g for 1 hour at 4°C, and the supernatant discarded. The exosome pellets were re-suspended in PBS buffer and used for detecting MICA expression with ELISA and Western blots.
Western Blots
Cell lysates were prepared by incubation in TNE buffer (20mM Tris-HCl pH 7.5, 150mM NaCl and 5mM EDTA) that contained 1% NP40 and protease inhibitors. After centrifugation to remove nuclei and cell debris, samples were run on 8% SDS-PAGE gels under reducing conditions (Genscript, cat M00812), and then were transferred to Amersham Hybond-P (GE Healthcare Life Sciences) with above exosome suspension. The membrane was blocked using 5% nonfat dry milk in TBS–0.1% Tween 20, and then MICAs were detected by incubating the membrane with MICA antibody (Abgent, Catalog: AP8626c) at 4°C overnight and then followed by anti-Rabbit horseradish peroxidase–conjugated secondary antibodies at room temperature for 2 hours. MICA proteins were visualized using SuperSignal West Femto Maximum sensitivity substrate (Thermo, Catalog: 34095).
RESULTS
Flow cytometry showed differential expression of membrane-bound MICA (Fig. 1), however intracellular MICA was similar for the three constructs suggesting that an equivalent amount of MICA protein was produced, but increased cell surface expression was associated with specific alleles. The mean fluorescent intensity and standard deviation (MFI/SD) of membrane-bound MICA*008, *007:01 and *019 in three experiments of the transfected fibroblasts were 4.5 +/-1.33, 6.71+/-0.4 and 9.86+/-2.0, respectively. Student’s t test showed significant p-values in comparisons between MICA*008 and *007:01 (p = 0.026), and MICA*008 and *019 (p = 0.018) (Fig. 1).
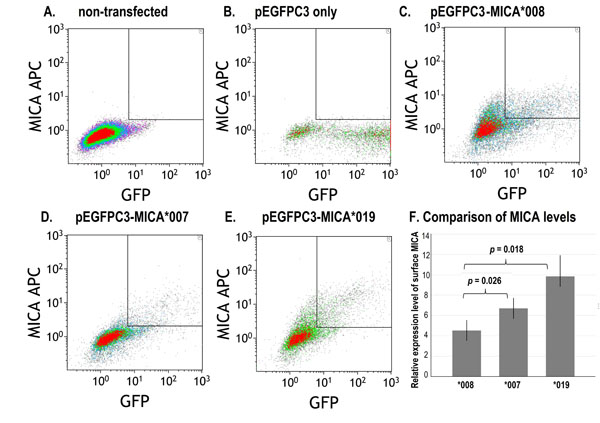
A representative dot plot of three flow cytometry studies showed differential expression of membrane-bound (surface) MICA*008, *007 and *019 in cultured fibroblasts. A. Fibroblasts without transfection; B. Fibroblasts transfected with pEGFPC3 without MICA; C. Fibroblasts transfected with pEGFPC3- MICA*008; D. Fibroblasts transfected with pEGFPC3-MICA*007; E. Fibroblasts transfected with pEGFPC3-MICA*019; F. Relative expression levels of surface MICA in three assays. MFI: mean fluorescence intensity. Error bars indicate standard deviations.
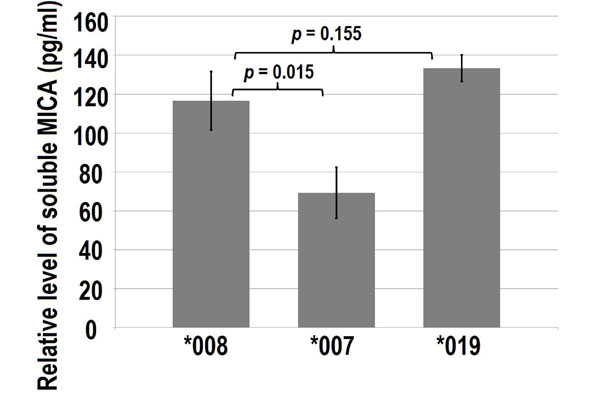
ELISA showing soluble MICA*008, *007 and *019 from cultured media of the fibroblasts (a total of 2.5 x105 fibroblasts in 5 ml culture media). Fibroblasts were transfected with pEGFPC3-MICA*008, pEGFPC3-MICA*007 and pEGFPC3-MICA*019. After 48-hours transfection, culture media were collected for MICA ELISA. Error bars indicate standard deviations.

Western blot showing exosomal MICA only detected in MICA*008 transfected fibroblasts. A total of 4 x106 fibroblasts were transfected with three MICA clones (MICA*008, *007 and *019). After 48- hours transfection, exosomes of each transfected fibroblasts were extracted and examined.
ELISA measurement for soluble MICA*008, *007 and *019 also displayed different levels (Fig. 2). Among the three examined MICA variants, MICA*007 showed significantly less soluble MICA. Comparing soluble MICA*008 verses *007:01 and MICA*008 verses *019.
ELISA measurement for exosomal MICA indicated a high level in the MICA*008 transfected fibroblasts. By contrast, MICA*007 transfected fibroblasts had a marginal level (minimal standard level) in only one of three tests, and MICA*019 exosomes were not detected. Further confirmation with Western blots showed that exosomal MICA only appeared in MICA*008 transfected fibroblasts (Fig. 3).
DISCUSSION
MICA expression usually occurs under cellular stress conditions, and it is involved in an immunosurveillance mechanism aiding elimination of infected or transformed cells [24, 25]. MICA controls the immune process through interaction with its receptor NKG2D. Expression of MICA on the cell surface (membrane-bound) activates NKG2D signaling. However soluble MICA (proteolytic product) and exosomal MICA (secreted form) down-regulate NKG2D [21-23]. Therefore, quantities of these three forms of MICA determine NKG2D mediated immune responses. Tumor-derived soluble MICA reduces NKG2D activity on CD8+ T and NK cells, and stimulates expansion of NKG2D+ CD4+ T cells with immune suppressor-like functions to evade NKG2D-mediated immune-surveillance [26]. By contrast, expression of MICA on the surface of synovial fibroblasts leads to up-regulation of MICA-NKG2D in local tissues that may cause auto-reactive T-cell stimulation, thus promoting the self-perpetuating pathogenic process in rheumatoid arthritis (RA) [27].
The studies herein compared expression of three MICA variants. We found that MICA*019 had the most cell surface expression followed by MICA*007, and MICA*008. This observation may reflect the level of activation of NKG2D signaling by each of these three MICA variants. Importantly, MICA*008 was the only variant found with an exosomal form, which together with its low presence on the surface of the fibroblasts may indicate that it has potentially a predominant inhibitory function on NKG2D signaling. By contrast, MICA*007 was found at the lowest level in soluble form among all three variants.
Three MICA variants displayed distinct expression on cell surface and proteolytic or exosomal forms suggesting their individual propensity in up- or down-regulation of NKG2D signaling. Therefore people carrying different MICA variants of these three alleles may respond to cellular stress with divergent paths after MICA-NKG2D mediated immune reactions. Previous genetic association studies indicated that the majority of AS patients carry MICA*007:01 and/or *019, while people carrying the most common MICA*008 allele showed significantly reduced risk of AS [1]. In addition, MICA*007 also confer susceptibility to UC [2], and MICA*019 to Behçet’s disease [3]. It is plausible to assume that patients who carry either MICA*007 or *019, and lack of MICA*008 may be more susceptible to an over-expression of MICA on the cell surface with greater activation of NKG2D leading to greater to cell-mediated cytotoxicity and inflammatory cytokine release from NK cells and T cells which is likely to produce more inflammation.
This is the first direct comparison of the expression of three MICA variants in primary human fibroblasts. The results of the studies together with previous reports of a high occurrence of MICA*007 and *019 in patients with chronic inflammatory diseases including AS, UC and Behcet’s disease indicated that over-presentation of these two MICA variants in patients may associate with a predominantly positive control for NKG2D mediated immune activation.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This study was supported by the NIH NIAID 1U01AI09090-01.

