All published articles of this journal are available on ScienceDirect.

Rituximab for the Treatment of Common Variable Immunodeficiency (CVID) with Pulmonary and Central Nervous System Involvement
Authors Info & Affiliations
Abstract
Background:
Granulomatous and lymphocytic interstitial lung disease (GLILD) represents a typical form of pulmonary manifestation of CVID. Except for glucocorticoid- and immunoglobulin-administration, no standardized treatment recommendations exist.
Objective:
To investigate our CVID-patients with GLILD for the applied immunosuppressive regimen, with a focus on rituximab.
Methods:
A retrospective analysis of all CVID-patients for the manifestation and treatment of GLILD at a single German center was performed in this study. For the evaluation of treatment-response, CT-imaging and pulmonary function testing were used.
Results:
50 patients were identified for the diagnosis of a CVID. 12% (n = 6) have radiological and/or histological confirmed diagnosis of a GLILD. Three patients received rituximab in a dose of 2 x 1000mg, separated by 2 weeks repeatedly. All patients showed radiological response and stabilization or improvement of the pulmonary function. Rituximab was used in one patient over 13 years with repeated treatment-response. Furthermore, the synchronic central nervous system-involvement of a GLILD-patient also responded to rituximab-treatment. With sufficient immunoglobulin-replacement-therapy, the occurring infections were manageable without the necessity of intensive care treatment.
Conclusion:
Rituximab might be considered as an effective and relatively safe treatment for CVID-patients with GLILD.
1. INTRODUCTION
Common variable immunodeficiency (CVID) represents a rare, heterogeneous primary immunodeficiency syndrome, which is characterized by hypogammaglobulinemia, impaired B cell differentiation, and poor or missing response to vaccination. These criteria must be fulfilled in a patient older than two years and any other causes for hypogammaglobulinemia must be excluded [1, 2]. CVID-patients are not only at the risk of infectious diseases but also of developing autoimmune and granulomatous manifestations [3-7]. This leads to the challenging situation of treating an already immunodeficient patient with an immunosuppressive regimen. Granulomatous and lymphocytic interstitial lung disease (GLILD) represents a common organ manifestation of CVID with a prevalence of up to 20% (8 - 22%) and is the type of lung involvement in CVID with the worst prognosis [8, 9]. According to the consensus statement of the British Lung Foundation and the United Kingdom Primary Immunodeficiency Network from 2017, GLILD is defined as a “distinct clinico-radio-pathological ILD occurring in patients with common variable immunodeficiency disorders, associated with a lymphocytic infiltrate and/or granuloma in the lung and in whom other conditions have been considered and where possible, excluded” [9]. Besides glucocorticoids, no standardized therapy of GLILD has been established yet [8, 9, 10]. In CVID-patients suffering from autoimmune cytopenia refractory to glucocorticoid-administration, rituximab-treatment has already shown convincing results [11-13]. Recently, there is growing evidence that rituximab, as monotherapy or in combination with antimetabolites (azathioprine or mycophenolate mofetil), represents an effective therapy for GLILD too [7, 14-25]. In this context, we want to provide our experience with three cases of CVID-patients who received rituximab for their GLILD. Furthermore, one of these patients had additional CNS-involvement by CVID, also responding to rituximab-treatment. CNS-involvement is an infrequent manifestation of CVID with lack of standardized treatment protocols [26-28].
2. METHODS
A retrospective analysis of our single-center database was performed for all patients with the diagnosis of CVID (inclusion and analysis up to March 2020). Ethical approval is not needed by German legislation due to its retrospective nature, as well as informed consent is not required due to anonymous data collection and storage. These patients were screened for GLILD as pulmonary manifestations of their CVID and for autoimmune cytopenia. All identified cases were then investigated for the applied immunosuppressive regimen for the treatment of GLILD. The use of rituximab was specifically investigated and evaluated for effectiveness in radiological response and pulmonary function testing, as well as for safety. To evaluate cytopenia as a risk factor for GLILD in our cohort, we performed a contingency table analysis and calculated the odds ratio (OR). As a test for significance, Fisher’s exact test was used and p < 0.05 was considered as significant. All statistical analysis was performed by Prism from GraphPad (Version 5.01, 7th August, 2007) (see supplemental data).
2.1. Classification According to the EUROclass Classification
The EUROclass classification divides CVID-patients into subgroups based on their B cell phenotype in flow cytometry and clinic manifestation [29]. Flow cytometry criteria for subdivision are the near absence of B cells (B: <1%), reduction of switched memory B cells (smB: less than 2%), expansion of CD21 low B cells (CD21: more than 10%), and expansion of transitional B cells (Tr: more than 9%) [29]. In granulomatous disease, a reduction of smB was discovered [29].
3. RESULTS
Currently, 50 patients with a diagnosis of CVID are followed up at our department of rheumatology and clinical immunology. While 32% (n = 16) have suffered from at least one form of autoimmune cytopenia, 12% (n = 6) of all CVID-patients have GLILD as a pulmonary manifestation (in four patients, a lung biopsy was performed, which revealed a pattern compatible with GLILD; two patients had only typical findings in CT imaging). In our patients with autoimmune cytopenia, GLILD was more frequent (31% vs. 3% in no-cytopenia patients), and the chance for development of GLILD was significant higher when patients suffered from cytopenia. (OR=15.00, 95% confidence interval 1.576 to 142.8, p = 0.0098). Regarding GLILD-treatment, one patient showed no progress under IgRT, and one patient was treated sufficiently with prednisolone only. Four patients needed further immunosuppressive treatment: three patients initially received azathioprine as a steroid-sparing agent, while one patient was primarily treated with rituximab. One patient reached stabilization of pulmonary function by azathioprine. However, one patient did not respond to azathioprine and developed additional myelotoxicity, and in another case, azathioprine had to be discontinued due to liver toxicity. These two patients then received rituximab-treatment. All cases of patients treated with rituximab are presented in detail below (Table S1-S3).
3.1. Case 1
Case 1 is a 35-year old female patient with an initial diagnosis of CVID at the age of 19 in March 2004. Genetic testing in 2018 revealed a gain of function mutation of STAT3. According to EUROclass classification, CVID was classified as B+SmB-CD21normTrnorm. IgRT was initiated in 2004. Pulmonary involvement accompanied by axillary and mediastinal lymphadenopathy, splenomegaly, thrombocytopenia, and leucopenia was diagnosed for the first time in 2005. Sampling from lymph nodes and bone marrow showed infiltration by a CD20+ lymphatic, non-malignant cell population. Video-assisted thoracic surgery (VATS) with lung biopsies from 2010 and 2013 confirmed the diagnosis of GLILD (see supplemental data). Due to IgRT-refractory immune thrombocytopenia with the need for high-dose prednisolone (> 20mg), rituximab-treatment (4 x 375mg / m2 IV) was started in September 2007. For addressing progressive pulmonary CD20+ lymphatic infiltrations, re-infusion of rituximab (2 x 1000mg absolute (abs) intravenous (IV)) in combination with prednisolone-treatment was performed in August 2010. A regression in lung manifestations and lymph node size was found in the re-assessment using CT imaging in November 2010 (Fig. 1). Recurrent pulmonary infiltration and renewed thrombocytopenia occurred during the course of the disease so that the re-administration of rituximab in August 2014, September 2015, June 2017, January 2019, and October 2019 (each in a dose of 2 x 1000mg abs IV) became necessary with continued treatment-response. From April 2014 until September 2019, the patient received additional azathioprine due to gastrointestinal manifestation. In summary, the patient received 7 cycles of rituximab. In CT-re-assessment in May 2019, no new infiltrates were detectable besides the constant chronic fibrotic parenchymal changes. In pulmonary function testing from 2007 until 2019, an improvement of FVC from 73% to 80% as well as of FEV1 from 63% to 69%, and stabilization of TLCO/VA at 60% was achieved. Despite starting rituximab-therapy in 2007, the patient had no major infections but developed recurring upper and lower respiratory tract infections with the necessity of frequent antibiotic treatments. A couple of times, hospitalization and IV-administration of antibiotics became necessary.
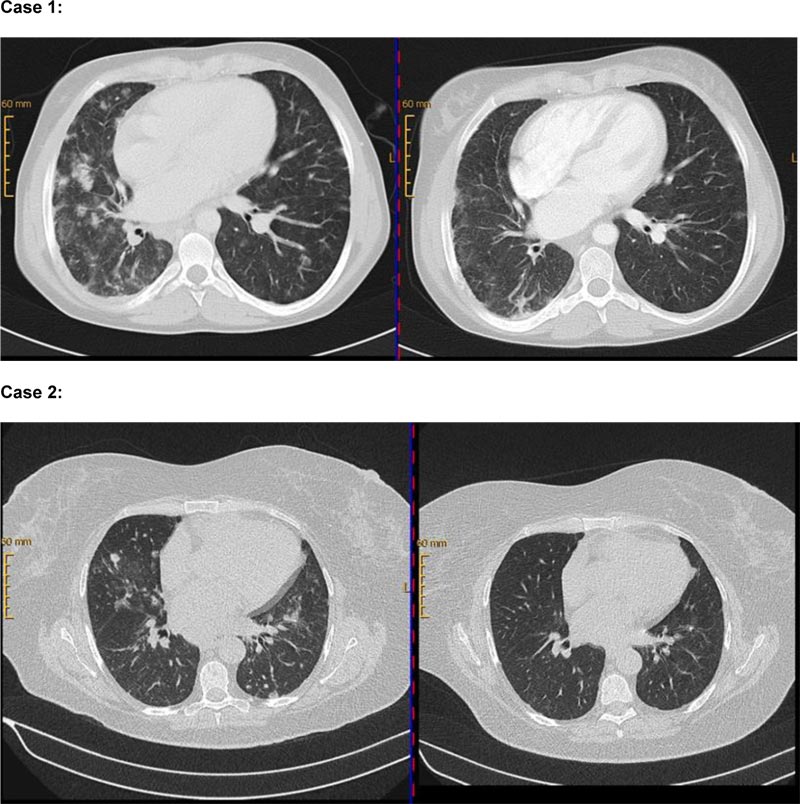
Lower row (Case 2): CT chest (08/2014) before rituximab-administration in 09/2014 (left side) and follow-up (right side) in 11/2014 with clearly perceptible regression of pulmonary manifestations.
3.2. Case 2
The second case represents a 66-year old female patient with a diagnosis of CVID in 2011. CVID was classified as EUROclass B+smB-CD21normTrnorm. In 2009, mediastinal, axillary and inguinal lymphadenopathy, splenomegaly, and pulmonary nodular infiltration were diagnosed for the first time by CT-imaging. Tissue sampling of lymph nodes, lung, and bone marrow showed infiltration by a CD20+ non-malignant B cell population and granulomas in the lung, respectively, so that GLILD diagnosis was confirmed (see supplemental data). Besides IgRT, prednisolone-treatment with 100mg was initiated in June 2013 due to pulmonary manifestation. No response could be achieved. A second prednisolone pulse accompanied by azathioprine was given in August 2014, when the patient developed autoimmune hemolysis and immune thrombocytopenia. Due to a lack of response to this treatment, rituximab (2 x 1000mg abs IV) was given in September 2014 for the first time. Azathioprine-treatment was stopped due to myelotoxicity. CT-imaging in November 2014 showed regression of pulmonary infiltration and lymphadenopathy (Fig. 1). In February 2017, the progress of pulmonary infiltration and renewed hemolysis led to rituximab-re-treatment (2 x 1000mg abs IV) in combination with a prednisolone pulse (250mg IV). Radiological re-assessment in July 2018, showed a good persistent response of the older lesions and also the development of new manifestations so that re-treatment is planned. Pulmonary function testing from 2013 until 2019 presented an improvement of FVC from 85% to 100% and of FEV1 from 96% to 100%, as well as of TLCO/VA from 92% to 103%. Besides a urinary tract infection, no bacterial infection necessitating antibiotic treatment occurred despite B cell depletion by rituximab. Furthermore, the patient discontinued IgRT for several months without having a bacterial infection.
3.3. Case 3
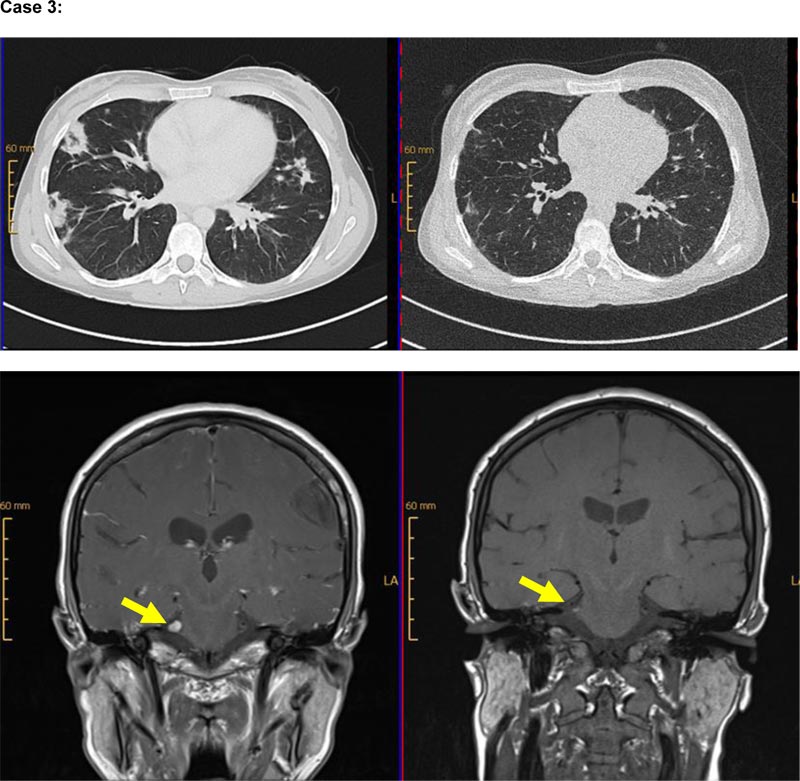
Our third case is a 40-year-old female patient with autoimmune hemolysis and immune thrombocytopenia, diagnosed with CVID in 2006 (classified as EUROclass B+smB-CD21normTrnorm). CT-imaging in 2007 revealed lymphadenopathy, splenomegaly, and pulmonary infiltration. IgRT was initiated in 2007. Progressive pulmonary infiltrates were detected, so a lung biopsy was performed in 2012 that confirmed the diagnosis of CD20+ GLILD (see supplemental data). Due to the lung involvement, the patient received prednisolone in a dose of 1mg/kg body weight. In 2014, new pulmonary manifestations were diagnosed by CT-imaging. Treatment with prednisolone was re-initiated, and azathioprine was added to the regimen. Azathioprine was discontinued in December 2015 because of liver toxicity. Due to relapsing pulmonary infiltrates, observed in imaging in June 2017, rituximab-therapy was administered for the first time in July 2017, and re-treatment was performed in January 2018 (each 2 x 1000mg abs IV). Excellent treatment response was confirmed by CT-imaging, showing the regression of pulmonary infiltrates in November 2017 (Fig. 2). In September 2017, the patient developed a sudden, short episode of aphasia and hypoesthesia of the hands. During the neurological assessment, lymphocytic pleocytosis (89 lymphocytes/ml) was seen in cerebrospinal fluid (CSF)-sampling without the detection of viral and microbial pathogens. Multiple small white matter lesions and one cortical lesion were found in cranial MR-Imaging (cMRI), due to which empiric anti-viral treatment was given. In August 2018, inflammatory CNS-symptoms relapsed with trigeminal hypoesthesia, lymphocytic pleocytosis in CSF-sampling (69 leukocytes/ml), and a corresponding t2-intense lesion in the cerebellopontine angle in cMRI accompanied by pulmonary progress in CT-imaging. Again, no infectious pathogen was found in CSF, so these CNS-involvements were retrospectively stated to be of autoimmune origin in the context of CVID. Rituximab re-treatment was given in September 2018 and achieved regression of cerebral and pulmonary manifestations confirmed by CT-imaging in December 2018 and cMRI in March 2019 (Fig. 2). In routinely assessed pulmonary function testing, an improvement of TLCO/VA from 71% to 80% could be achieved, as well as the stabilization of FVC at 110% and of FEV1 at 105%. During treatment with rituximab, the patient suffered from some minor infectious diseases, including respiratory tract infections like sinusitis or bronchitis. Only one pneumonia occurred as a single major infection over the years under B cell depletion, but it was manageable without the necessity of intensive care treatment.
4. DISCUSSION
In this case series, we provide our experiences in treating GLILD-patients with a particular focus on the use of rituximab. When immunosuppressive therapy became necessary, we usually initiated prednisolone-treatment. In case of insufficient response or relapse, patients received additional azathioprine. Due to lack of response or intolerance of azathioprine, patients then received rituximab in a dose of 2 x 1000mg abs IV, separated by two weeks. The three female patients who presented with generalized lymphadenopathy, splenomegaly, autoimmune cytopenia and GLILD, and additional CNS-involvement in one case were treated repeatedly, efficiently, and safely with rituximab. All three patients’ CVID was defined as B+smB-CD21normTrnorm [29]. Diagnosis of GLILD was confirmed radiologically and histologically in all three cases corresponding to the definition provided by the British Lung Foundation and the United Kingdom Primary Immunodeficiency Network [9]. Regarding safety concerns, no severe infectious diseases occurred despite the B cell depletion. All occurring infections were manageable, and no patient developed septical complications with the necessity of intensive care treatment.
Our experience contributes to the already existing knowledge of the management of GLILD and the effective use of rituximab in this condition. Two larger studies have already evaluated the different treatment options in GLILD: A recently published online survey by the e-GLILDnet presented the experiences of 161 centers and physicians of 47 different countries [25]. Besides this largest study on the same topic, an older retrospective analysis on 32 patients of the French DEFI cohort with different granulomatous organ involvements was carried out by Boursiquot et al. [7]. Both recommend glucocorticoids as first-line treatment to induce remission, as we also did for our patients [7, 25]. There are several recommended options for tapering glucocorticoids. Antimetabolites like azathioprine or mycophenolate mofetil, rituximab, or combinations of rituximab and an antimetabolite represent the most common treatments [25]. Due to the suspected crucial role of B cell hyperplasia in the pathogenesis of GLILD, B cell depletion seems to be a promising treatment strategy [30]. Most evidence exists for the use of rituximab in combination with an antimetabolite. Several case reports, such as a case series of seven patients carried out by Chase et al. and a recently published larger study on 39 patients from Verbsky et al., reported convincing treatment response [14, 15, 19, 24]. We performed, mainly, rituximab-monotherapy, which was also described in several case reports as an effective therapy before [16-18, 20-22]. In the large e-GLILDnet survey, physicians reported the best treatment outcomes in patients receiving either rituximab-monotherapy or a combination of rituximab and mycophenolate mofetil [25]. Nevertheless, most of the participant centers in the survey recommended rituximab or mycophenolate mofetil, prior to the combination therapy of both, as the best option [25].
While in most of the published literature, rituximab was administered in a dose of 4 x 375mg/m2 every week for 4 weeks [14-17] [20-22, 24]. We administered rituximab successfully in a dosage of 2 x 1000mg abs, separated by two weeks, which offers the benefit of a lower total dose, lower costs, and lower hospitalization rates. Additionally, due to the relapsing and heterogeneous nature of GLILD, we did not use a protocol with fixed re-infusion intervals but guided our therapy based on the clinical necessity. As described by Verbsky et al., B cell regeneration after rituximab correlates with GLILD relapse and might be used as a follow-up parameter after rituximab-therapy, besides CT-imaging and lung function testing [24]. When GLILD relapsed, our patients repeatedly responded to rituximab re-treatment so that the total dose of immunosuppression could be minimized in these already immuno compromised patients by this procedure. The prevalence of autoimmune cytopenia was 32% in our cohort, slightly higher than that reported in other studies [3, 4]. GLILD seemed to be more frequent in our CVID-patients with cytopenia, and cytopenia represented a significant risk factor for developing GLILD in our analysis. Rituximab is an effective treatment for this condition in CVID due to which this subgroup of patients suffering from both, GLILD and autoimmune cytopenia, might benefit twice from B cell depletion [11-13].
Randomly controlled interventional studies for the treatment of autoimmune and granulomatous manifestations in CVID are not available due to the fact that CVID is a heterogeneous disease with a very low prevalence of 1: 25000 [31]. The evidence for management has to be deducted from retrospective analyses like this case series. Therefore, our case series might provide additional evidence for the effectiveness and safety of rituximab-treatment in CVID with organ involvement.
CONCLUSION
In the CVID-case with additional and relapsing CNS-involvement, rituximab in combination with prednisolone, primarily administered due to pulmonary manifestation achieved a clinical and cMRI-morphological regression of the neurological manifestation. Non-infectious CNS-involvement represents an infrequent condition in CVID for which prednisolone-treatment is recommended [7, 28]. For tapering of glucocorticoid-dose or managing glucocorticoid-refractivity, no evidence is available due to this extremely rare manifestation. Our case report suggests that rituximab might be effective in the treatment of CNS-involvement.
KEY MESSAGES
- Rituximab might be considered as an effective and relatively safe long-term treatment of CVID-patients with GLILD.
- Administration of the 2 x 1000mg dose seems to be effective in our patients.
- Larger treatment-trials with more patients are necessary and seem to be promising for gaining further evidence for the effectiveness of rituximab in GLILD.
- A case of CNS-manifestation responded to rituximab-treatment.
LIST OF ABBREVIATIONS
| CVID | = Common Variable Immunodeficiency |
| GLILD | = Granulomatous and Lymphocytic Interstitial Lung Disease |
| IV | = Intravenous |
| abs | = Absolute |
| FVC | = Forced Vital Capacity |
| FEV1 | = Forced Expiratory Volume |
| DLCO | = Diffusing Capacity |
| IgRT | = Immunoglobulin Replacement Therapy |
| VA | = Alveolar volume |
AUTHORS' CONTRIBUTION
PPS and MS collected the data, PPS, ECS, and MS interpreted the data, MS supervised the study, PPS, MF, MG, ECS, LKN, AK, HPT, and MS wrote the paper, MF, MG, ECS, LKN, AK, HPT, and MS approved the final version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABLITY OF DATA AND MATERIALS
The authors confirm that the data supporting the findings of this study are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers web site along with the published article.

