All published articles of this journal are available on ScienceDirect.

A Case Report of Successful Treatment of Recalcitrant Childhood Localized Scleroderma with Infliximab and Leflunomide
Abstract
Herein we report successful treatment of an adolescent Caucasian female with severe progressive localized scleroderma (mixed subtype, including generalized morphea and linear scleroderma of the trunk/limb) using infliximab and leflunomide. The patient demonstrated improvement after the first 9 months of therapy based on her clinical examination, objective measures, and patient and parent global assessments. Infliximab is a potential treatment option for pediatric localized scleroderma patients who have progression of disease or who are unable to tolerate the side effect profile of more standard systemic therapy. Larger longitudinal studies or case series are needed to confirm and further investigate infliximab’s role in localized scleroderma.
INTRODUCTION
Localized scleroderma (LS) affects approximately 1-3 per 100,000 children with a typical onset of 7 – 9 years of age. Since LS is an uncommon condition, it is under recognized and there is often a significant lag period from the development of symptoms to diagnosis and treatment initiation (average 2-3 years) [1]. The main clinical subtypes of LS include linear scleroderma (trunk/extremity and head), generalized morphea, circumscribed morphea (superficial and deep) and mixed morphea [2]. In children, the most common subtype is linear scleroderma (50-60%), which has the potential for the most disability secondary to its effect on joints and resultant contractures from deep tissue involvement [1]. A recent survey of the pediatric LS subjects included in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) registry demonstrated that 28% of the 259 children with LS had functional limitation due to musculoskeletal changes, such as muscle atrophy and limb length discrepancy [3]. A longitudinal cohort study confirms this disability persisting into adulthood 30 years later [4].
The pathophysiology of localized scleroderma is characterized by a lymphocytic infiltration into the perivascular area with fibroblast activation that causes thickening of the collagen bundles, decreased elasticity of the fascia, and skin thickening over time. The clinical effects of these changes can be both cosmetic and functional depending on the involved area. Some clinical manifestations that are particularly concerning are lesions of the head (Parry-Romberg Syndrome, En coup de sabre) thatcause obvious structural changes to the face but can also be associated with dental, brain and eye abnormalities, linear lesions of the extremities overlying joints that cause a limitation of the joint function and may progress to contracture, and deep lesions infiltrating the extensor and flexor tendons associated with finger and hand function [1, 2]. Systemic treatment is geared toward halting the progression of the disease and allowing the patient to regain any lost function through skin remodeling in conjunction with non-pharmacologic modalities such as physical and occupational therapy.
The Childhood Arthritis and Rheumatology Research Alliance (CARRA) released consensus treatment plans as a guideline for the treatment of pediatric localized scleroderma in 2012 [5]. These general recommendations include treating moderate to severe LS with corticosteroids (CS) (either enteral or parenteral dosing is appropriate) and methotrexate (subcutaneous formulation is preferred). While these guidelines acknowledge that other treatments may be needed, the only other treatment discussed and agreed upon by experts in the field at the time was the use of mycophenolate mofetil (MMF) for recalcitrant cases or intolerability to methotrexate (MTX).
CASE
A 14 year old Caucasian female presented to our pediatric scleroderma clinic with a 4 year history of progressive mixed morphea, a combination of generalized plaque morphea and linear scleroderma of the trunk/ extremities. Her condition started as a single bruise-like lesion over the right flank, and despite systemic therapy, had advanced to cover approximately 80% of her body surface area.
A deep punch biopsy of the initial lesion demonstrated histologic findings consistent with LS (morphea), including full thickness dermal fibrosis with a superficial and deep perivascular lymphohistiocytic infiltrate. In addition, there was thickening of the fibrous septa in the panniculus with aggregates of plasma cells at the edges, supporting deeper involvement. An eosinophilic infiltrate was not present in the subcutis. She was initially treated with topical corticosteroids, but these were discontinued due to their ineffectiveness to halt disease with expansion of lesions noted. Oral treatment was initiated, though not considered as standard therapy, with penicillin VK and hydroxychloroquine (400mg daily) (Fig. 1). Approximately 1 year later she was started on more traditional steroid and methotrexate therapy, initially with oral prednisone, followed by IV methylprednisolone administered per Toronto protocol (1000 mg IV daily three consecutive days) [6]. Due to intolerability of the methotrexate (maximal dose 17.5 mg subcutaneous weekly), she was switched to mycophenolate mofetil (MMF) and continued on it for another year at a maximum dose of 1500 mg twice daily. She started to complain of odd side effects with MMF, including migraine headache and short term memory loss, leading to its eventual discontinuation. She received another course of monthly three consecutive days of 1000 mg daily IV methylprednisolone for 3 months prior to our evaluation in clinic (Fig. 1).

Timeline of medical therapy. A graphic summary of the past 5 years of treatment for the patient reported in the case. Topical corticosteroids (Top CS), Penicillin VK (Pen VK), Hydroxychloroquine (HCQ), Prednisone (Pred), Methotrexate (MTX), Minocycline (MNC), Mycophenolate mofetil (MMF), Infliximab (IFX), Leflunomide (LEF), IV Methylprednisolone (MP) and No treatment (No Tx).
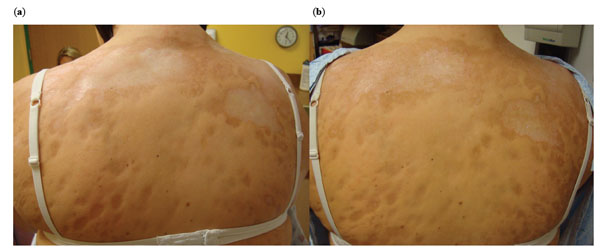
Pre- and Post-treatment photographs of the upper back. The initial back lesions (a, left panel) demonstrate yellow/white waxy lesions with thick skin coalescing over her upper back. Post-treatment photographs (b, right panel) demonstrate decreased white/yellow waxy appearance of lesions, with less uniform skin thickness and ‘breaking up’ of lesions.
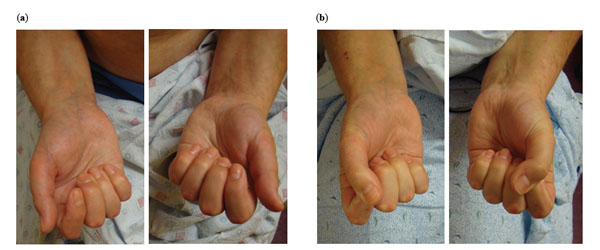
Pre- and Post-treatment photographs of the hands. (a) Pre-treatment photographs (left panel) demonstrate diffuse swelling of fingers and limited range of motion. The patient was unable to make a fist. (b) Post-treatment photographs (right panel) demonstrated marked decrease in swelling and the ability to close her fist.
The history obtained from the patient and documentation from the treating physician at the time of our initial consultation supported a partial response to both oral prednisone and IV methylprednisolone pulses, characterized by less stiffness and slightly improved joint mobility. Other therapies were not thought to contribute to a significant change in disease course (above). In the months prior to evaluation in our clinic she continued to show continued disease progression, characterized by appearance of new, expanding, and thickening of lesions. Her review of systems was otherwise unremarkable for signs of systemic sclerosis, including absence of Raynaud phenomenon, or other overlapping connective tissue disease features apart from signs of upper GI irritation while on prednisone.
On our initial examination, she demonstrated a variety of findings, including both active disease and damage features. Most notably were two overall cutaneous and deep tissue patterns. One consisted of diffuse large coalescing plaque morphea lesions covering her trunk, with mild erythematous borders, yellow/white waxy indurated centers and moderate to severe skin thickness throughout. The other consisted of deep linear bands of thick skin traveling down the majority of her upper and lower extremities, concentrated on the extensor surfaces, spreading out and affecting the digits with diffuse limitation range of motion of both large and small joints. An associated general edema was also demonstrated of her peripheral extremities. Please see pre-treatment photographs in Figs. (2, 3) and pre- infliximab treatment durometer readings (skin hardness) and joint function in Table 1.
Comparison of clinical outcome measures pre- and post-infliximab treatment.
| Clinical Outcome | Pre-Infliximab Treatment | Post-Infliximab Treatment |
|---|---|---|
| Cutaneous outcome measures: | ||
| mLoSSI (range: 0 – 216) | 39 | 22 |
| LoSDI (range: 0 – 162) | 93 | 84 |
| Durometer reading (durometer units): | ||
| Upper back- left | 24- 35 | 18- 20 |
| Upper back- right | 25- 38 | 25- 33 |
| Joint function (all from right side*): | ||
| Upper Extremity | ||
| 3rd finger (cm)** | ||
| Extension FTP distance | 9.7 | 10.0 |
| Flexion FTP distance | 2.0 | 0.7 |
| Delta Finger-to-palm (FTP) | 7.7 | 9.3 |
| Elbow | 25- 50% limitation | 0% limitation |
| Lower Extremity | ||
| Hip | < 25% limitation | 0% limitation |
| Knee | < 25% limitation | 0% limitation |
| Ankle | 50 – 75% limitation | 50 – 75% limitation |
| Subtalar | 50 – 75% limitation | 25 – 50% limitation |
| Global Assessment of Function: | ||
| Physician | ||
| PGA- Activity | 45 | 0 |
| PGA- Damage | 75 | 45 |
| PGA- Severity | 80 | 53 |
| Patient/Parent | ||
| Patient Global Severity | 40 | 2 |
| Parent Global Severity | Not completed | 0 |
| CDLQI (range 0-30) | 9 | 6 |
mLoSSI: modified localized scleroderma skin severity index, a composite score to reflect cutaneous disease activity.
LoSDI: localized scleroderma damage index, a composite score to reflect cutaneous disease damage components.
CDLQI: Childhood Dermatology Life Quality Index; higher score indicated higher impact on QOL.
* Both the left and right side affected equally, right represented for simplicity.
** When involvement of all fingers, such as in this patient, the 3rd finger is typically used as the representative finger for finger-to-palm (FTP) measurements, as used in systemic sclerosis.
Her initial laboratory investigation included a negative ANA (due to this negative value no other extractable nuclear antigens were tested at presentation), and was only remarkable for a mild eosinophilia with absolute count of 630 and mild elevation in the erythrocyte sedimentation rate, 34mm/hr (normal ≤ 20mm/hr). The C-reactive protein level was normal. Subsequent laboratory evaluation showed the development of a positive ANA at 1:320 titer with negative values for routine extractable nuclear antigens, including Scl-70. The single stranded DNA antibody, which is associated with more severe LS, was positive at 130 U/mL (normal value ≤ 69).
Infliximab was considered in our patient due to medication intolerance and disease progression during the use of standard LS therapies, MTX, MMF and corticosteroids. A review of the literature found a few cases of infliximab's efficacy in adults with generalized morphea (no pediatric cases had been reported) and its effect on skin improvement in both limited and diffuse cutaneous forms of systemic sclerosis [7-9]. Infliximab (10mg/kg) was administered to our patient as monthly infusions. She was followed locally by another pediatric rheumatology provider (PW) and within the first 2 infusions her fingers and ankles felt less full/stiff and skin less thick on her trunk. Four months into infliximab therapy, leflunomide was started to supplement therapy and to decrease the likelihood of formation of auto-antibodies to infliximab. She continued with physical and occupational therapy, and in conjunction with systemic therapy, allowed her to regain a large amount of lost function. Her additional medications included a proton-pump inhibitor and an anti-histamine. She did not have any serious complications with therapy, although she developed a herpetic mouth lesion 9 months into infliximab therapy which required treatment with oral valacyclovir prior to resolution.
At a follow-up clinic visit at our center, 9 months after the initiation of infliximab therapy, the patient reported a significant improvement in the range of motion of almost all her joints and marked improvement of her grip strength. Table 1 reports the improvement in the joint and skin findings post-infliximab. She noted that her skin felt less thick, which was confirmed in several areas on physical examination using the modified Rodnan skin score assessment (0-3 scale). The improvement was further supported by the comparison of durometer readings (skin hardness) for several lesions, pre- and post-treatment (Table 1). In general, on examination the thick/indurated waxy lesions broke up into smaller islands of decreased skin thickness and the erythematous halo had resolved in regard to the large plaque morphea type of lesions. The linear lesions affecting the upper and lower extremities still had areas with moderate to severe thickness within the bands, but these were narrow and more isolated to the center of the lesion compared to prior. The general edema of the distal upper and lower extremities greatly diminished and the overall ROM of large and small joints improved (see Table 1 and Fig. 3).
Our clinic uses a comprehensive (and validated) skin scoring technique that includes the modified localized scleroderma skin severity index (mLoSSI) to reflect cutaneous disease activity [10] and the localized scleroderma damage index (LoSDI) to reflect cutaneous disease damage components [11], which together compose the localized scleroderma cutaneous assessment tool (LoSCAT). Other prospectively collected clinical outcomes measured are standardized joint range of motion (ROM) charting (including finger to palm distance) [12], the Childhood Dermatology Life Quality Index (CDLQI) [13] and global assessment of disease severity on a 10cm VAS scale from the perspective of the parent, the child and the treating physician [10]. In general, other objective modalities are employed to monitor responsiveness to therapy in LS include imaging (MRI, Ultrasound, thermography) and non-imaging techniques, such as the computerized skin score [14], skin biopsy and durometer measures. In our clinic, we will commonly employ the durometer in tandem assessment to clinical cutaneous scoring [15]. The durometer is an engineering tool developed to assess the hardness of materials and has been used to assess the hardness of sclerodermatous skin for several years. A study in 2008 found that durometer readings have excellent correlation with disease activity in systemic sclerosis and are helpful in giving an objective measure for multiple skin points [16]. Additionally, they noted that the variance in durometer readings correlates well to the patient’s experience and frequently there are improvements in durometer changes readings that correlate with the patients perception [16].
In our patient, there was significant improvement in almost all scores (cutaneous, joint and global assessment) after 9 months of treatment with infliximab (Table 1). Of note, both accepted measures of disease activity in LS, the mLoSSI and PGA-A [10], improved markedly by reaching pre-determined minimal clinically important difference scores (6 and 41, respectively) [17], confirming a truly significant clinical change in disease status.
DISCUSSION
As this patient has had several different courses of medications, including standard regimens of methotrexate, corticosteroids (CS) (both oral and intravenous) and MMF with continued progression of disease, the therapeutic challenge was to determine which immunomodulator would give the greatest efficacy with the least intolerable side effect profile to this patient.
Infliximab is a monoclonal antibody directed against soluble and cell-surface bound TNF-α. It is FDA approved for use in inflammatory bowel disease, ankylosing spondylitis, psoriasis, psoriatic arthritis, and rheumatoid arthritis. It is also frequently used off-label for many conditions in pediatric rheumatology, such as juvenile idiopathic arthritis, uveitis, and less commonly for dermatomyositis and mixed connective tissue disease, typically with dramatic and sustained results on joint and eye inflammation [18-20]. Systemic sclerosis (SSc) and localized scleroderma (LS) are thought to be ‘companion’ diseases sharing similar histologic inflammatory and fibrotic components despite some clinical phenotypic differences. The pathogenesis of SSc is related to fibrocyte activation and collagen hypertrophy signaled from infiltrating lymphocytes and resident macrophages. TNF-α is a key driver of communication between T cells, macrophages, and fibrocytes and it is postulated that inhibiting this communication would reduce the pathologic process. TNF-α staining in the affected skin of a patient with severe generalized plaque morphea was demonstrated by Diab et al. [7]. After treatment with infliximab, this LS patient showed marked clinical response in parallel with decreased TNF-α staining in the skin on repeat biopsy [7].
Our patient had a very protracted course of disease, which unfortunately spanned over many years and resulted in severe damage. Her escalating disease course and an uncommon side effect profile of her medications warranted a more non-conventional approach as it was reflected by using multiple systemic medications. At the time of this report she is maintained on both infliximab as monthly infusions and a daily dose of leflunomide. The low dose leflunomide was prescribed intending to reduce the possible formation of neutralizing auto-antibodies against infliximab. All providers feel that it is the infliximab that provided the most impact on improvement, which was noted even prior to addition of leflunomide, and represents a viable choice for recalcitrant localized scleroderma not responding to traditional medications or in cases of intolerance to these therapies.
CONCLUSION
Localized scleroderma in children is associated with severe morbidity due to the deleterious effect on the function of the involved area but also due to the emotional impact associated with long term cosmetic changes. While systemic treatment with methotrexate, corticosteroids, and mycophenolate mofetil are available and often effective, these may be associated with side effects that are intolerable to the patient or they may not completely control disease progression. Our patient felt that the regimen with infliximab and leflunomide was tolerable and greatly improved her function. The validated measurement tools mentioned above also confirmed that she was making excellent progress supporting the continuation of her current treatment plan. To our knowledge, we report the first successful use of infliximab for treatment of recalcitrant pediatric localized scleroderma. We hope that this report may serve as a sentinel case for development of further studies of TNF-α inhibitors’ effect on this disease.
AUTHORS CONTRIBUTION
Dr(s). Ferguson, Torok and Weiser had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Acquisition, analysis, and interpretation of the data: Ian D. Ferguson, MD, Peter Weiser, MD, Kathryn S. Torok, MD
Drafting of the manuscript: Ian D. Ferguson, Peter Weiser, Kathryn S. Torok.
Critical revision of the manuscript for important intellectual content: Ian D. Ferguson, Peter Weiser, Kathryn S. Torok.
CONFLICT OF INTEREST
Ian D. Ferguson is employed in fellowship by the University of Pittsburgh and Children's Hospital of Pittsburgh of UPMC and has no other financial relationships.
Peter Weiser is employed by Children’s of Alabama, Birmingham, AL and holds a volunteer assistant professor position at the University of Alabama at Birmingham, AL.
Kathryn S. Torok is dually employed by the University of Pittsburgh and the Children’s Hospital of Pittsburgh of UPMC and has no other financial relationships.
ACKNOWLEDGEMENTS
Ian D. Ferguson is supported in his fellowship by National Institute of Health (T32, AR052282).
Peter Weiser has no grant support.
Kathryn S. Torok is supported by the following grants: Nancy Taylor Foundation for Chronic Diseases Inc. and the NIAMS Mentored Patient Oriented Research Award, NIH Grant No. K23 AR059722
There were no sponsors associated with this report: the design; data collection, analysis, and interpretation; preparation, review of the manuscript and decision to submit the manuscript for publication were all completed by the authors.

