All published articles of this journal are available on ScienceDirect.

Signal Transduction Pathways in Chronic Inflammatory Autoimmune Disease: Small GTPases
Abstract
Ras superfamily small GTPases represent a wide and diverse class of intracellular signaling proteins that are highly conserved during evolution. These enzymes serve as key checkpoints in coupling antigen receptor, growth factor, cytokine and chemokine stimulation to cellular responses. Once activated, via their ability to regulate multiple downstream signaling pathways, small GTPases amplify and diversify signaling cascades which regulate cellular proliferation, survival, cytokine expression, trafficking and retention. Small GTPases, particularly members of the Ras, Rap, and Rho family, critically coordinate the function and interplay of immune and stromal cells during inflammatory respones, and increasing evidence indicates that alterations in small GTPase signaling contribute to the pathological behavior of these cell populations in human chronic inflammatory diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Here, we review how Ras, Rap, and Rho family GTPases contribute to the biology of cell populations relevant to human chronic inflammatory disease, highlight recent advances in understanding how alterations in these pathways contribute to pathology in RA and SLE, and discuss new therapeutic strategies that may allow specific targeting of small GTPases in the clinic.
INTRODUCTION
Investigators conducting pioneering histological evaluations of rheumatoid arthritis (RA) patient synovial tissue were struck by the gross similarities between hyperplastic synovium and solid tumors found in cancer patients, especially in regard to the “tumor-like proliferation” of stromal fibroblast-like synoviocytes (FLS) localized to the synovial intimal lining layer [1, 2]. Subsequent in vitro analyses of isolated RA FLS further extended the conceptual association of synovial hyperplasia with oncogenic transformation, as FLS displayed many of the phenotypic characteristics inexorably linked to cancer cells. RA FLS display enhanced proliferative capacity compared to FLS obtained from healthy tissue or patients with other forms of arthritis, the cells can proliferate in an anchorage-independent manner in vitro without inhibition by cell-cell contact, and constitutively secrete autocrines and matrix metalloproteinases (MMPs), supporting proliferation and tissue invasion [3-5]. In a poignant extension of these similarities, elegant studies have demonstrated that activated RA FLS can migrate from collagen implants in mice to unaffected joints and initiate inflammation and cartilage destruction at distal locations, a phenomenon markedly analogous to tumor cell metastasis in cancer [6]. These intrinsic properties of RA FLS are not transient responses to inflammatory stimuli. Instead, it is clear that this phenotype is imprinted upon RA FLS, as gene expression profiles of RA patient synovial tissue and FLS cultured ex vivo from the same patients are highly similar [7]. This imprinted phenotype has important functional consequences, as the invasive properties of RA FLS in vitro highly correlate with the rate of joint destruction occurring in the patients from which they were obtained [8, 9].
The molecular mechanisms contributing to this “semi-transformed” phenotype of RA FLS are still poorly understood. One of the possibilities initially explored was that early inflammatory events in the synovium might generate somatic mutations in proto-oncogenes or tumor suppressors, altering FLS behavior [10]. These first studies understandably focused on genes at the forefront of cancer biology, but attempts to identify somatic mutations in primary candidates such as ras genes or the tumor suppressors phosphatase and tensin deleted on chromosome 10 (PTEN) or p53 were either unsuccessful or failed to support a general and supporting role for such mutations in conferring an imprinted aggressive phenotype to RA FLS [11-16]. Thus, while a simple molecular mechanism responsible for the “semi-transformed” phenotype of RA FLS continues to elude us, studies addressing this question have promoted a rapid increase in the identification and understanding of intracellular signal transduction pathways, persistently activated by constant exposure of synovial cells to a complex inflammatory cytokine and cell-cell milieu, that are important to pathology in RA and other chronic inflammatory diseases [17]. Not surprisingly, many of these intracellular signaling pathways are critically regulated by the same proto-oncogene and tumor suppressor gene products important in cellular transformation. In this review, we will examine recent advances in our understanding of how members of the Ras superfamily of GTPases contribute to immune-mediated inflammatory diseases, and highlight evolving opportunities for targeting these signaling proteins therapeutically.
RAS SUPERFAMILY GTPASES
The Ras superfamily of small GTPases constitutes a group of more than 100 structurally related proteins which regulate a large spectrum of cellular processes, ranging from acute responses to extracellular stimuli, to roles commonly described as “housekeeping functions” [18]. Ras superfamily GTPases can be classified into one of five families based on structural similarities, the Ras, Rho, Rab, Ran, and Arf families, as well as the “orphan” GTPases Miro1, Miro2, and RhoBTB3. In very general terms, members of each family regulate distinct cellular processes. Ras family GTPases couple extracellular stimuli to regulate cellular gene transcription, proliferation, survival and integrin activity, while Rho family GTPases couple the same stimuli to regulation of gene expression and cytoskeletal organization. Rab and Arf family GTPases control receptor internalization and intracellular vesicular trafficking, while Ran GTPases are responsible for microtubule stability and cargo transport between the cytoplasm and nucleus [19]. Roles for Miros and atypical Rho GTPases are just being explored [20, 21]. Structurally, small GTPases consist of a phosphate-binding loop, responsible for binding guanosine diphosphate (GDP) and guanosine triphosphate (GTP), flexible “switch” regions, which mediate binding to downstream effector proteins, and unique carboxy-terminal tails, modified by the addition of farnesyl or geranylgeranyl moieties, regulating subcellular localization of the GTPase. Cellular stimuli can activate guanine nucleotide exchange factors (GEFs), which displace GDP from specific small GTPases. Freed from GDP, the small GTPase quickly binds GTP, which induces a conformational change allowing the GTPase to associate with and activate multiple downstream signaling proteins. Hydrolysis of bound GTP to GDP, catalyzed by specific GTPase-activating proteins (GAPs), returns the GTPase to its inactive state [22]. Mutations that abolish or diminish susceptibility to GAPS (e.g., glycine 12 to valine in Ras; RasV12) result in constitutive activation of the small GTPase, while mutations that decrease dissociation from GEFs, preventing binding to GTP (e.g., serine 17 to asparagine in Ras; RasN17) result in dominant-negative molecules which block activation of endogenous GTPases.
RAS FAMILY GTPASES, RAS REGULATORY PROTEINS, AND THEIR EFFECTOR PATHWAYS
The closely related Ras family GTPase homologues (H-, K-, and N-Ras) are expressed throughout mammalian tissue, play important roles in coupling extra-cellular stimuli to multiple downstream signaling pathways, and, at least during development, have a high degree of functional redundancy. While no readily evident phenotypes are observed in mice lacking H-Ras, N-Ras or the K-Ras4A splice variant, alone or in combination, genetic deletion of both splice variants of K-Ras results in embryonic lethality [23, 24]. The ability of many of the Ras homologues to functionally compensate for each other during development likely arises from their highly shared sequence identity, especially in the effector domain coupling GTPases to downstream signaling proteins. Activation of Ras proteins is regulated by three major groups of Ras GEFs, the mammalian son-of-sevenless (mSos) proteins, Ras guanine nucleotide releasing proteins (RasGRPs) and Ras guanine nucleotide-releasing factors (RasGRFs) (Fig. 1) [25]. mSos is recruited to activated receptor tyrosine kinase or non-receptor tyrosine kinases via adaptor proteins, resulting in localization of mSos at the plasma membrane. This allows mSos interaction with membrane phospholipids via its pleckstrin homology (PH) domain, and subsequent activation [26, 27]. RasGRPs are activated to varying degrees by intracellular calcium and/or binding to diacyl glycerol (DAG) or other phorbol esters, while the molecular mechanisms by which RasGRFs are activated have yet to be fully elucidated [25, 28-31]. Amongst Ras family GEFs, RasGRF1 appears to have the highest selectivity in vivo, preferentially activating H-Ras but not K-Ras, N-Ras or the more distally related R-Ras [32, 33]. Well-characterized downstream signaling pathways of Ras family proteins include serine/threonine kinase cascades leading to activation of the mitogen-activated protein (MAP) kinases p38, extracellular-regulated kinase (ERK), and c-jun N-terminal kinase (JNK), phosphatidylinositol 3-kinases (PI3-Ks), and Ral family GEFs (Fig. 1) [34, 35]. Specificity in Ras homologue signaling is conferred in part by differential subcellular localization of each homologue. Unique carboxy (C)-terminal sequences and differential post-translational modifications of the C-terminal peptide direct localization of Ras homologues to distinct cellular membrane compartments, and regulate the inclusion or exclusion of Ras homologues from lipid rafts [36]. This in turn impacts upon the ability of each Ras protein to interact with distinct GEFs and downstream effector proteins [37, 38]. Details of MAP kinase and PI3-K involvement in chronic inflammatory autoimmune disease, and developing efforts to target these enzymes therapeutically are discussed elsewhere in this journal series.

Regulation of RA FLS activation by Ras family GTPases. In RA FLS, the H-Ras –specific GEF RasGRF1 is over-expressed. Furthermore, unidentified cellular proteases cleave RasGRF1, resulting in its constitutive activation. This leads to H-Ras activation, promoting high basal transcription of IL-6 and MMP-3. Exposure of RA FLS to inflammatory cytokines leads to further activation of H-Ras, as well as inducing activation of K-Ras and N-Ras. Through their combined and redundant activation of MAP kinase and PI3-kinase signaling pathways, H-, K-, and N-Ras each contribute to RA FLS chemokine, cytokine, and MMP production that perpetuates inflammation and promotes disease progression.
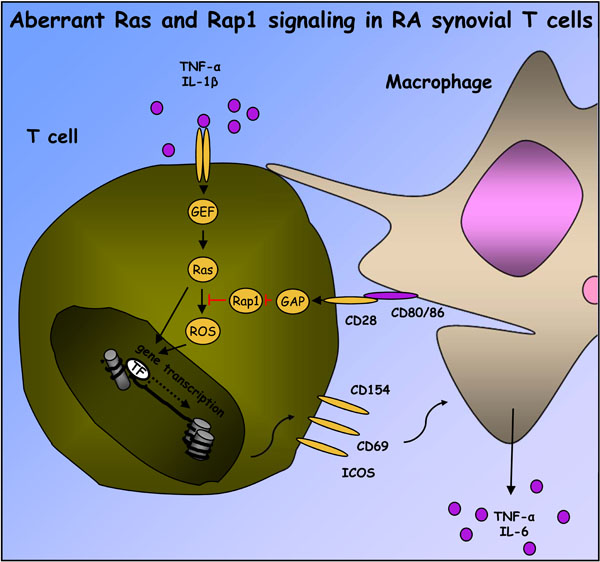
Contributions of Ras and Rap signaling to RA synovial T cell activation. Exposure of T cells to IL-1β and TNFα, among other inflammatory cytokines, leads to activation of unidentified T cell Ras GEFs and Ras proteins. ROS-dependent and –independent pathways downstream of Ras can stimulate T cell inflammatory gene transcription. Ras-dependent signaling in T cells is usually buffered, directly or indirectly, by activation of Rap1. However, in RA synovial tissue, macrophage CD80 and/or CD86 stimulate RapGAP activity associated with CD28, preventing activation of Rap1. In the absence of Rap1 signaling, T cell ROS and other Ras-dependent signaling pathways are enhanced, promoting expression of ICOS, CD154 and perhaps other activation markers, which in turn perpetuate activation of synovial macrophages.
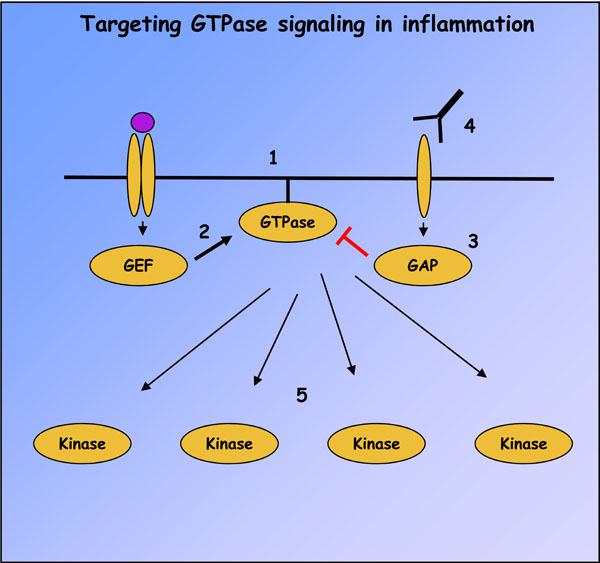
Strategies for targeting small GTPase signaling in immune-mediated inflammatory diseases. Ras superfamily GTPase signaling can be abolished with compounds such as FTIs, geranylgeranyl transferase inhibitors and inhibitory peptides which prevent GTPase membrane localization (1). GTPase activation can also be modulated by small molecular weight compounds targeting GEF catalytic activity or interactions with GTPases, of which activators of EPAC and inhibitors of DOCK2 and Trio are the first examples (2). Similar compounds have not yet been developed to target GAP catalytic activity (3), but receptor antagonists may be identified which can indirectly modulate GAP activity, as in the case of CTLA4-Ig and RapGAP (4). Finally, many downstream effectors of GTPases, like MAP kinases and PI3-Ks, possess catalytic activity and represent pharmacological targets (5).
GENETIC MANIPULATION OF RAS SIGNALING AND IMMUNE PHENOTYPES
Genetic analyses of fibroblasts from mice lacking H-Ras or N-Ras indicate that these two GTPases regulate different gene profiles [39]. Lack of N-Ras induces expression of a number of genes involved in innate immune responses and promotion of apoptosis, while down-regulating adhesion molecules and promoters of cell cycling [39, 40]. Redundancies in H-Ras and N-Ras –dependent gene expression have also been noted in H-Ras-/-/N-Ras-/- double knockout animals. Here, basal and transforming growth factor β1 -induced synthesis of extracellular matrix components is enhanced in fibroblasts lacking both GTPases, associated with suppressed ERK activation and enhanced PI 3-kinase signaling [41].
Examining immune responses in vivo, mice lacking Ras GEFs and downstream components of the Raf/ERK kinase cascade have severe blocks in both positive selection of thymocytes, and T cell antigen receptor (TCR) responses of peripheral T cells [42]. In N-Ras knockout mice, a partial defect in the positive selection of only CD8+ thymocytes is observed [39, 43]. These mice are more susceptible than their wild-type counterparts to primary influenza infection, likely due to decreased T cell interleukin (IL)-2 production following antigen stimulation. However, as is the case with many TCR-proximal signaling components, such as ZAP-70, partial inactivation of T cell Ras signaling pathways during development not only interferes with positive selection and peripheral T cell antigen-responsiveness, but, counter-intuitively, induces autoimmunity [44]. Mice lacking RasGRP1, which in addition to mSos is the major GEF responsible for activating Ras proteins following TCR ligation, display a characteristic decrease in the number of mature CD4+ and CD8+ thymocytes, decreased peripheral T cell numbers and proliferative responses, and an inability to clear bacterial and viral infections [45-47]. TCR-dependent Ras activation in T cells from these mice, though detectable, is greatly reduced. Despite this, CD4+ T cells which make it to the periphery adopt an activated memory phenotype, and induce systemic autoimmune disease characterized by age-dependent lymphadenopathy, splenomegaly, and auto-antibody production characteristic of SLE [46]. Ras, as well as the related GTPase Rap1, is activated during both positive and negative selection, and disease in RasGRP1-deficient mice likely arises from the inability of weakened Ras signaling to transmit pro-apoptotic signals which would otherwise negatively select thymocytes during encounter of high-affinity self-antigens [46, 48]. Indeed, direct analyses of TCR β-chain repertoires in RasGRP1-deficient mice under homeostatic conditions shows a pronounced skewing of TCR utilization compared to that found in wild-type (WT) mice [49].
RAS FAMILY SIGNALING IN RA AND SLE
Initial immunohistochemical studies indicated that Ras proteins were expressed in RA synovial tissues, but drew differing conclusions regarding whether Ras expression was elevated in RA synovial tissue compared to disease controls and healthy individuals [5, 50]. Recent analyses with homologue-specific antibodies have indicated that similar levels of both K-Ras and N-Ras proteins are found in the synovial tissue of RA patients compared to patients with psoriatic arthritis (PsA), reactive arthritis, and inflammatory osteoarthritis (OA), while H-Ras protein and mRNA expression is selectively elevated in RA, especially in FLS. H-Ras, K-Ras and N-Ras are all expressed at highly variable levels in RA FLS, and each of the homologues is activated following FLS stimulation with IL-1β or TNFα [51]. Basal activation of H-Ras is readily detectable in unstimulated RA FLS in vitro, likely due to over-expression and activating post-translational modification of RasGRF1 [52]. The activation status of Ras proteins in synovial tissue has not been directly measured, as affinity probes which can precipitate specific activated small GTPases have not proven useful in immunohistochemical studies, and the degree of overlap in Ras homologue signaling pathways precludes the use of surrogate biomarkers to infer the activation status of Ras proteins in synovial tissue. However, one or more of the Ras proteins is constitutively activated in freshly derived RA synovial fluid T cells, likely due to stimulation by multiple inflammatory cytokines present in synovial fluid [53].
Numerous independent studies have provided evidence that Ras proteins play an important role in RA FLS activation, and contribute to pathology in animal models of RA. Farnesyltransferase inhibitors (FTI), which prevent proper membrane localization of Ras proteins, some Rho family GTPases, and signaling components of G protein-coupled receptors, decrease the incidence and severity of collagen-induced arthritis (CIA) in mice, accompanied by decreased synovial TNFα and IL-1β mRNA expression [54, 55]. FTI also inhibit TNFα-induced MMP-1 production in RA FLS, accompanied by decreased activation of JNK and NF-êB signaling pathways [56]. Over-expression of a dominant-negative c-Raf kinase in RA FLS, which broadly binds to and inhibits Ras homologues and related Ras family members, suppresses growth factor-induced activation of ERK and JNK MAP kinases, interferes with FLS MMP production, and slows FLS proliferation and invasiveness in vivo [57, 58]. Presumably more selective interference with H-Ras signaling, via ectopic expression of dominant-negative H-Ras, suppresses IL-1β -induced ERK activation and IL-6 production in RA FLS, and suppresses joint destruction in experimental arthritis [59]. Additional evidence for a specific role for H-Ras in RA is derived from studies indicating that RasGRF1 is highly expressed in RA synovial tissue and FLS, and detected in a cleaved form thought to result in its constitutive activation in RA [52]. RasGRF1 expression in RA synovial tissue co-localizes and correlates with MMP-3 production, and in vitro contributes to constitutive MMP-3, IL-6 and IL-8 production by RA FLS [52]. Interestingly, protease-dependent cleavage of RasGRF1 was previously identified as an important mechanism for driving H-Ras activation and invasiveness in melanoma cancer cell lines, drawing yet another striking similarity between RA FLS and cancer cell phenotypes [60, 61].
Contributions of distinct Ras family members to cellular function in RA synovial tissue are now being addressed through more systematic approaches. Although it is likely that all Ras homologues are activated in RA synovial fluid T cells, persistent reactive oxygen species (ROS) production observed in these cells can be mimicked in Jurkat T cells by transfection of active H-Ras, but not K-Ras or N-Ras [53, 62]. In this experimental system, H-Ras –induced ROS production requires the downstream small GTPase Ral, and a cell-permeable inhibitor of H-Ras can block synovial fluid T cell ROS production. In RA FLS, each of the Ras homologues contributes to autonomous cellular activation, as well as cellular responses to IL-1β [51]. Consistent with effects observed during manipulation of RasGRF1 expression levels in RA FLS, transfection of RA FLS with active H-Ras, but not K-Ras or N-Ras, enhances basal MMP-3 production. However, active mutants of each of the Ras homologues can enhance basal IL-1β-induced MMP-3 production. For IL-8, active mutants of Ras protein can enhance basal production, but only active H-Ras cooperates with IL-1β to further stimulate IL-8 expression. In reciprocal experiments silencing Ras homologue expression in RA FLS, H-Ras was found to be required for basal IL-6 and MMP-3 production, while N-Ras activity was required for IL-8 expression. Each of the Ras homologues contributed to IL-1β -induced IL-6 production, while selective requirements were observed for H-Ras and N-Ras in the induction of MMP-3 and IL-8 respectively [51]. Expression of active K-Ras has also been observed to enhance RA FLS proliferation in vitro, although this effect can be suppressed by TNFα or TGFβ [63]. These data would collectively suggest that although specific signaling attributes can be identified for each Ras homologue in FLS activation, each Ras protein contributes to global parameters mediating inflammation and joint destruction. In keeping with this, use of pan-Ras locked nucleic acid (LNA) to simultaneously knockdown all three Ras homologues in vivo showed superior efficacy to N-Ras LNA in protecting mice against CIA. One caveat to this conclusion, however, is that synovial analysis of Ras family mRNA expression following pan-Ras LNA treatment indicated that, locally, H-Ras expression was most efficiently targeted by the pan-Ras LNA [51]. Together, this data suggests a scenario in RA synovial tissue FLS where constitutive activation of H-Ras by RasGRF1synergizes with inflammation-dependent activation of K- and N-Ras to promote FLS activation (Fig. 1).
The first indication that Ras family GTPases may contribute to cellular activation in SLE was reported in a pioneering observation from Klinman and colleagues, where they noted that expression of N-Ras, among other proto-oncogene products, was over-expressed in peripheral blood mononuclear cells of SLE patients [14]. Other investigators have also identified decreased expression of the Ras family GEF SOS in SLE patient PBMCs, associated with constitutively high activation of cellular kinase activity attributed to ERK1 and ERK2 MAP kinases [64, 65]. More recent studies to detect specific MAP kinase activation have provided evidence that activation of ERK and JNK, but not p38, in SLE patient PBMCs correlates with disease activity [66]. In contrast to bulk PBMCs, T cells from SLE patients are defective in their ability to mount a proliferative response, associated with an inability to integrate TCR signaling with CD28 and ICOS costimulatory pathways [67]. Defective ERK activation following SLE T cell stimulation may be attributed in part to defective coupling of SOS to the adaptor protein Grb2 [68]. Intriguingly, a study of a relatively small group of normal donors and SLE patients identified 13 novel splice variants of RasGRP1 in PBMCs, many of which encoded defective transcripts [69]. Expression of these defective RasGRP1 transcripts was enhanced in SLE PBMCs, and in T lymphocytes, RasGRP1 expression was often diminished or undetectable. Again, although a causal role remains to be established for altered Ras signaling in SLE pathology, this last molecular observation is striking given the lupus-like phenotype of RasGRP1-deficient mice [45, 46, 49, 70]. No studies have yet examined the effects of targeting other Ras family signaling components in animal models of SLE, but administration of FTI does suppress disease in lupus-prone mice, again suggesting involvement of Ras or related proteins [71].
RAP FAMILY GTPASES, RAP REGULATORY PROTEINS, AND THEIR EFFECTOR PATHWAYS
The Rap family consists of Rap1 (a and b) and Rap2 (a and b), each of which is encoded by distinct genes but share 95% amino acid identity [72]. In over-expression systems, it is difficult to discern differences between Rap proteins in their ability to regulate cellular functions, but as discussed below, targeted disruption of individual genes in vivo is clearly defining roles for Rap1a and Rap1b in distinct cell populations. Rap GEFs are activated by a wide range of protein-protein interactions, as well as by soluble second signaling messengers. These GEFs include C3G (similarly to mSOS, recruited to tyrosine-phosphorylated receptors and complexes via the SH2 domain of an associated adaptor protein), postsynaptic density 95/disc-large/zona occludens (PDZ)-GEFs I and II, exchange proteins directly activated by cAMP (EPAC) 1 and 2, and calcium and diacylglycerol regulated GEF (CalDAG-GEF, RasGRP2). Two groups of GAPs regulate inactivation of Rap proteins, the RapGAP (RapGAP1A, RapGAP1B, and RapGAP2) and Spa-1 (Spa-1, Spa-1-like, and E6TP1) families [22]. The best-characterized function of Rap1 proteins is the regulation of adhesion proteins in cells of both hematopoietic and non-hematopoietic origin. Rap1 protein activation mediates integrin activation, integrin-dependent chemotaxis, cadherin-mediated adhesion, and cell-cell junction formation, likely via the activation of multiple coordinated signaling pathways. One downstream effector of Rap1 proteins important in integrin-dependent adhesion is Riam, an adaptor protein which in turn interacts with actin regulatory proteins, talin, ADAP (adhesion- and degranulation-promoting adaptor protein), and SKAP-55 (Src kinase-associated phosphoprotein of 55 kDa) [73-75]. Active Rap1 can also associate with RAPL (regulator of adhesion and cell polarization enriched in lymphoid tissues, also known as Nore1B and Rassf5), an adaptor protein which couples Rap1 to and activates the serine-threonine kinase mammalian sterile twenty-like-1 (Mst1) to induce LFA-1 clustering [76, 77]. In vitro studies have also indicated that afadin (AF-6) is a Rap1 effector, which inhibits endocytosis of E-cadherin and allows the maintenance of cellular junctions [78-80]. Finally, Rap1 can directly couple to and regulate multiple GEFs and GAPs for Rho family GTPases, providing a link between integrin adhesion and cytoskeletal remodeling [81].
GENETIC MANIPULATION OF RAP SIGNALING AND IMMUNE PHENOTYPES
The study of a large number of genetically-modified mice strains has collectively identified specific roles for Rap1a and Rap1b in lymphocyte activation and trafficking, innate immunity, angiogenesis and maintenance of vascular integrity. Initial studies focused on the role of Rap1a in T cell biology. Transgenic over-expression of constitutively active Rap1a-E63 in the T cell compartment of mice confers constitutive LFA-1 integrin activation to thymocytes and peripheral T lymphocytes. In these mice, reduced CD4+ T lymphocyte proliferative responses to anti-CD3 antibodies as well as recall antigens, and T cell help for B cell Ig class switching are observed [82]. Although suppressed CD4+ Th cell proliferative responses in Rap1a-E63 transgenic mice are to some extent cell-intrinsic effects, the numbers and immunosuppressive capacity of FoxP3+ regulatory T cells (Tregs) is also increased. In these mice, Rap1a-E63 promotes development of naturally occurring Tregs by LFA-1 integrin-independent mechanisms, although integrin activation partially contributes to Rap1-induced differentiation of inducible Tregs [83]. Intriguingly, transgenic expression of another active Rap1a mutant, Rap1a-V12, in T cells only partially phenocopies Rap1a-E63 mice [84]. In contrast to Rap1a-E63, which is a constitutively active mutant lacking intrinsic GTPase activity that is insensitive to GAPs, Rap1a-V12 is a hypermorphic mutant displaying an approximately 200 –fold decrease in sensitivity to GAPs [22]. Additionally, unlike Rap1a-E63, which is over-expressed compared to endogenous Rap1a, Rap1a-V12 is expressed at levels comparable to endogenous Rap1. As a result, T cell Rap1a activity levels in Rap1a-V12 transgenic mice are comparable to those achieved by phorbol ester and ionomycin stimulation of endogenous Rap1a [84]. Thymocytes and T cells from Rap1a-V12 transgenic mice do display constitutive integrin activation, but no impairment in TCR/CD28 –induced ERK activation or proliferation [84]. Additionally, Rap1a-V12 has no influence on TCR/CD28-induced IL-2, IFNγ, or TNFα production, nor the development or induction of Tregs and Th17 cells [85]. This may suggest that many of the phenotypes observed in Rap1a-E63 are consequences of non-physiological Rap1a signaling. However, mice lacking Spa-1, a RapGAP enriched in hematopoietic tissues, particularly in proliferating T lymphocytes, display an age-dependent decrease in T cell proliferative responses to CD3/CD28 and recall antigen stimulation, and are unable to generate effective antibody responses [86]. Later in life, these mice also generate auto-reactive B cells [87].
Genetic deletion of Rap1a has no overt effect on B or T cell development, nor lymphocyte proliferative responses to antigen receptor ligation. However, lymphocyte integrin function is modestly reduced, demonstrating both a physiological role for Rap1a in integrin activation, as well as likely functional redundancies with Rap1b [88, 89]. An independent study of Rap1b knockout mice has indicated that Rap1b is the major Rap1 protein expressed in T cells, but potential impacts of Rap1b on T cell function have not been reported [90]. To date, the quality of T cell immune responses in vivo in mice lacking Rap1a or Rap1b have not been examined. However, mice transgenically expressing RapGAP1 in the T cell compartment display an age-dependent accumulation of activated T cells in spleen and lymph nodes, and these T cells are hyper-responsive to mitogenic stimulation [91]. Rap1b knockout mice display a strong B cell phenotype, demonstrating a required role for Rap1b in maintaining integrin function during B cell development, homing, and T cell interactions in vivo [92]. Numbers of B220+IgM- pro/pre –B cells and B220+IgM+ immature B cells are reduced in Rap1b –deficient mice, as are marginal zone B cells in the spleen. Proliferative responses of B cells are unaffected, as are T cell-independent humoral responses, while T cell-dependent antibody responses are greatly reduced. Intriguingly, recent evidence has emerged that signals generated by Rap1 through RAPL bifurcate into pathways regulating both cellular adhesion and cell cycling [93]. Although young RAPL-/- mice are lymphopenic, aged mice display lymphoproliferative disease, associated with the development of lupus-like antibody production, and B cell lymphomas. Initial studies indicate that in addition to regulating integrin activation, RAPL couples antigen receptor stimulation to suppression of kinase interacting stathmin (KIS) -dependent phosphorylation and nuclear localization of the cell cycle regulator p27kip1.
Rap1 signaling is also important for myeloid cell homing and function in vivo. Integrin-dependent adhesion to fibronectin and vitronectin is reduced in macrophages from mice lacking Rap1a, and macrophages from these mice demonstrate increased random movement on adhesive matrices [88]. Fc-R-mediated phagocytosis is slightly increased in Rap1a-deficient macrophages, while oxidative bursts are decreased. Differences between Rap1a-deficient and WT macrophages are relatively moderate, again indicative of functional redundancies between Rap1a and Rap1b. Downstream of Rap1, RAPL is essential not only for T and B cell homing in vivo, but also the migration of dendritic cells to spleen and draining lymph nodes during immune challenge [94, 95].
Lastly, Rap1 function appears to play a key role in angiogenesis and vascular endothelial integrity. Rap1a-deficient mice demonstrate reduced neovascularization following hind limb ischemia, while decreased retinal neovascularization is observed in Rap1b-deficient mice [92, 96]. Decreased blood vessel formation, induced by VEGF, bFGF, and FGF2, is observed in Matrigel plugs implanted into mice lacking either Rap1a or Rap1b, and endothelial cells derived from these mice display decreased integrin activation, migration, and permeability in vitro [92, 96, 97]. At least in vitro, Rap1 regulation of neovascularization is partially dependent upon signaling to RAPL [92, 97].
RAP FAMILY SIGNALING IN RA
The role of Rap1 signaling in RA has been investigated primarily within the context of T cell contributions to pathology. Initial indications that Rap1 activation may contribute to T cell anergy, coupled to studies reporting that RA synovial T cells were unresponsive to TCR signaling, suggested the possibility that Rap1 may be aberrantly activated in RA synovial T cells. Suprisingly, analysis of Rap1 activation status in RA synovial fluid T cells revealed a complete block in CD3 and phorbol ester/ionomycin –induced Rap1 activation [53]. Subsequent studies found that inactivation of RA synovial fluid T cell Rap1 was mediated by the T cell costimulatory protein CD28. Synovial fluid T cell Rap1 inactivation could be mimicked in RA patient peripheral blood T cells incubated with autologous synovial fluid macrophages, or peripheral blood-derived macrophages exposed to synovial fluid, in a cell contact-dependent manner [98]. Previous work had already demonstrated that TCR-dependent activation of Rap1 could be efficiently blocked by CD28 costimulation, and inclusion of CTLA4-Ig (Abatacept) in T cell-synovial fluid macrophage co-cultures prevented Rap1 inactivation (Fig. 2) [91, 98, 99]. Maintenance of Rap1 signaling by CTLA4-Ig was associated with a failure of T cells to produce high levels of ROS, and transduction of T cells with Rap1a-V12 prior to their incubation with synovial macrophages also blocked subsequent T cell ROS production [98]. ROS are proposed to act as important second messengers in T cell activation, and scavenging intracellular ROS has been shown to suppress TCR-induced NF-κB, AP-1 and IL-2 promoter transcription [100]. Reciprocally, chronic T cell oxidative stress can lead to constitutive activation of NF-κB -dependent inflammatory gene products [101].
Together, these studies indicated that restoration or maintenance of Rap1 function in T cells might alleviate their pathological contributions to RA. Consistent with this, Rap1a-V12 mice are highly resistant to clinical manifestations of CIA [85]. Rap1a-V12 mice show decreased disease incidence and severity, and undetectable levels of cartilage and bone damage. Protection against CIA in Rap1a-V12 mice is also associated with decreased anti-collagen autoantibody production. The precise cellular and molecular mechanisms by which sustained T cell Rap1 activity protects mice against CIA are currently unknown, but their delineation will be necessary in establishing the potential of T cell Rap1 modulation as a therapeutic strategy in established RA. It is unlikely that Rap1a-V12 expression impacts on the TCR repertoire during selection to remove auto-immune T cells, as previous characterization of these mice found little evidence of Rap1 effects on positive and negative selection [84]. Rap1a-V12 also fails to induce T cell anergy in the CIA model, as TCR/CD28 proliferative and cytokine responses in healthy Rap1a-V12 mice are normal [84, 85]. Moreover, during the onset of CIA, the distribution and number of effector/memory T cells is identical in Rap1a-V12 and WT mice, and Rap1a-V12 splenic and lymph node T cells make expected levels of IL-2, IFNγ, IL-17 and IL-10. Also, Rap1a-V12 mice behave identically in terms of clinical responses, viral clearance, antibody production, and CD4+ and CD8+ T cell homing and cytokine responses during primary and secondary influenza infection (our unpublished observation). Intriguingly however, there is a specific defect in Rap1a-V12 lymph node T cell production of TNFα [85]. This might be due to clonal exhaustion of autoimmune T cell clones, a phenomenon previously observed in murine antigen-specific T cell populations during chronic viral infection [102, 103]. Secondly, under highly inflammatory conditions, Rap1a-V12-mediated integrin activation may prolong T cell contact with antigen-presenting cells, leading to activation-induced cell death. Such an effect of T cell Rap1 activation has previously been observed in vitro [104]. Finally, the specific lack of TNFα production may be due to the inability of Rap1a-V12 T cells to up-regulate costimulatory expression needed for TNFα production. In vitro, Rap1a-V12 T cells from healthy mice show a modest decrease in ICOS and CD154 expression following TCR/CD28 stimulation [85]. Engagement of these costimulatory proteins is important not only for T cell TNFα production, but is also required for follicular T cell help that promotes B cell immunoglobulin class switching [105, 106]. It remains to be established whether Rap1a-V12 T cells also fail to induce ICOS and CD154 expression during CIA in vivo. However, available data suggests a model wherein constitutive activation of Ras proteins by synovial inflammatory cytokines, in combination with macrophage-dependent CD28 costimulation of T cells and Rap1 inactivation, leads to synovial T cell ROS production and expression of activation markers which further stimulate macrophages (Fig. 2).
Despite a large community interest in the role of integrins and cadherins in the migratory and invasive properties of RA FLS, little data is available regarding Rap1 function in these cells. Recently, an initial study has identified a potential role for cAMP-dependent activation of Rap1 by prostaglandin E2 in RA FLS [107]. However, the physiological consequences of Rap1 activation in RA FLS remain to be identified.
RHO FAMILY GTPASES, THEIR REGULATORY PROTEINS, AND EFFECTOR PATHWAYS
Rho family GTPases are best known for their role as key regulators in coupling extracellular stimuli to maintenance and reorganization of cytoskeletal structure, as well as cell-cycle progression, and MAP kinase-dependent gene expression [108]. The mammalian Rho family is comprised of 20 members, four of which, Rac1, Rac2, RhoA and Cdc42, have been best characterized. Rho GTPases have been shown to control cellular motility and polarity in migrating cells by regulating actin and myosin organization. In this context, Rac1 is often associated with lamellipodium formation and membrane ruffling, as well as regulation of the oxidative burst, Cdc42 with actin microspike and filopodium formation, and RhoA with the promotion of actin stress fiber formation and focal adhesion assembly [108]. Rho family GTPases also often play a more prominent role in the activation of serine/threonine kinases leading to p38 and JNK activation than Ras GTPases [109]. Human Rho family GTPases are regulated by a bewildering array of GEFs and GAPs. Some 70 Rho family GEFs are comprised of two families of proteins, the Dbl homology (DH) family, and DOCK family, which can be activated by receptor tyrosine kinases (or receptor-associated tyrosine kinases), G-coupled protein receptors, integrins and other receptors [110, 111]. Genome sequencing has indicated that up to 70 different RhoGAPs may be expressed in humans, only a handful of which have been well-characterized [112]. Bound to GTP, Rho family GTPases can interact with a large number of effector proteins including not only those that directly regulate cytoskeletal rearrangements, but also couple to PI 3-kinase, MAP kinase, and NF-êB signaling pathways [108].
GENETIC MANIPULATION OF RHO FAMILY SIGNALING AND IMMUNE PHENOTYPES
A number of mice deficient in the expression of Rho family members, such as Rac1, Rac2, Cdc42, and RhoH have been generated, and each displays immune phenotypes [113]. Complete deletion of Rac1 results in embryonic lethality, but use of targeted deletion strategies has revealed that many cellular functions of Rac1 in the immune system are redundantly regulated by Rac2 [113, 114]. Deletion of either Rac1 or Rac2 alone has no impact on T cell development, but simultaneous deletion of both GTPases blocks early thymocyte development, prior to mature TCR expression, as well as positive selection [115-118]. In mature T cells, Rac2 is required for chemotactic responses in vitro and in vivo, and may contribute to TCR-dependent proliferative responses, Th1 maturation and IFNγ production, although this requirement is not strong enough to affect Th1-dependent responses to infection [115, 116, 119]. In B cells, Rac1, together with Rac2, plays an important role in BCR-induced activation, transducing BCR signals that control survival and cell cycle entry [120, 121]. Unique roles for Rac1 and Rac2 appear in myeloid lineage cells. Deletion of either Rac1 or Rac2 in neutrophils decreases chemotactic responses in vitro and in vivo, but only Rac2 is required for NADPH oxidase function [122]. Further analyses have demonstrated that Rac1 regulates neutrophil polarization toward chemotactic agents, while Rac2 is responsible for motoric migration towards the chemoattractant [123]. Rac1 is required for macrophage cell spreading and membrane ruffling, but neither Rac1 nor Rac2 appear to be required for efficient macrophage migration [124, 125]. A specific required role for Rac2 has been demonstrated in macrophage oxidative bursts and Fcγ receptor-mediated phagocytosis [126]. Dendritic cells (DCs) lacking expression of both Rac1 and Rac2 show defective cytoskeletal rearrangements, migration, antigen presentation and formation of immunological synapses with T cells, preventing adequate T cell priming [127]. Finally, in osteoclastogenesis, both Rac1 and Rac2 contribute to osteoclast development in vitro. Rac1 is selectively required for M-CSF-dependent migration of pre-osteoclasts and RANKL-induced superoxide bursts. These specific attributes of Rac1 signaling in osteoclastogenesis are also evident in vivo, as mice in which Rac1 is conditionally deleted in myeloid lineage cells, but not Rac2-deficient mice, show increased trabecular bone volume [128]. Lineage-specific requirements for Rac protein signaling are also observed in mice deficient for DOCK2, a Rac-specific GEF expressed in hematopoietic cells. B and T lymphocytes and plasmacytoid DCs, but not monocytes or myeloid DCs, from DOCK2-deficient mice display defective chemotactic responses in vitro, and lymphocytes fail to home to lymphoid organs in vivo [129, 130]. Neutrophils from these mice do respond to chemoattractants, but are hampered in the speed of their response [131].
Knockout mice have been generated for a number of the Rho GTPases, which have in particular revealed requirements for these proteins in regulating T cell development and function. During thymocyte development, functional Rho is required through the early pre-T cell stage for cell survival, while in late pre-T cells, Rho activity is needed for thymocyte proliferation [132]. Additionally, Rho activation is sufficient and necessary for thymocyte integrin-dependent adhesion and chemotactic responses [133]. In mature cytotoxic T cells and natural killer cells, each of the components of the RhoA/Rho kinase (ROCK)/LIM-kinase pathway is required for cytoskeletal arrangements critical for cell-mediated killing [134]. Roles for both RhoA and RhoB have been identified in MHC II surface expression and antigen-dependent activation of T cells by DCs [135, 136]. RhoH, expressed specifically in hematopoietic lineage cells, is required for proper TCR signaling in thymocytes and mature T cells but suppresses chemokine-dependent adhesion [137-139]. However, Rho function is not restricted to T lymphocytes, as mast cells from RhoH-deficient mice display defects in degranulation and cytokine production [140].
Deletion of Cdc42 signaling components in vivo has also demonstrated specific roles for this GTPase in immune development and responses. Mice lacking Cdc42GAP display elevated levels of Cdc42-GTP in bone marrow-derived neutrophils [141]. In vitro, random movement of these neutrophils is enhanced, while directed movement in response to fMLP is reduced. In the presence of integrin ligand and fMLP, enhanced Cdc42 activity promotes ERK activation while suppressing p38 MAP kinase activation, and the effects of Cdc42 activation on random and directed movement, is dependent upon Cdc42 signaling to these MAP kinases [141]. Specific deletion of Cdc42 in the B cell lineage significantly reduces development of mature B cells [142]. Mature B cells which do develop in these mice produce reduced IgM, IgG1 and IgG3 and are insensitive to BAFF receptor signaling, but do show normal chemotactic responses. Requisite roles for Rho, Rac, and Cdc42 proteins in osteoclast development and activation have been well-characterized, and we refer the reader to a recent comprehensive review on this subject [143].
RHO FAMILY SIGNALING IN RA AND SLE
As evidenced by genetic studies, Rho family proteins would be predicted to make important contributions to the recruitment and activation of many cell populations involved in initiating inflammation and bone erosion in RA. Rac1 is constitutively activated in cultured RA FLS, and exposure of RA FLS to NSC23766, a small-molecular weight inhibitor of Rac1-3, or silencing of Rac1 expression, blocks their proliferation but does not cause apoptosis [144]. NSC23766 also reduces RA FLS invasiveness in vitro, in large part through suppressing Rac-dependent JNK activation. Little is known about the role of Rac GTPases in animal models of arthritis. Rac2-deficient mice also lacking Rac1 in their myeloid compartment have been examined in Chlamydia-induced arthritis. Here, lack of Rac1 and Rac2 protects against clinical signs of arthritis in the early stages of disease, but exacerbates disease during the chronic stage [145]. Due to the inability of Rac1 and Rac2 -deficient neutrophils to induce TLR4 in response to Chlamydia, initial inflammatory signaling during the acute phase of disease is tempered, while later, the lack of clearance of infection exacerbates arthritis. Rac1 has also been targeted pharmacologically in CIA, using a cell-permeable Rac1 C-terminal peptide fragment which specifically blocks signalling of Rac1 but not other Rho family GTPases [146]. In a prophylactic model of CIA, Rac1 inhibitory peptide has no effect on disease incidence, disease severity, synovial infiltration by inflammatory cells, cartilage erosion, or bone destruction. Significant decreases in paw swelling are observed, possibly due to the ability of Rac1 inhibitory peptide to prevent disruption of endothelial vascular integrity and subsequent edema. Anti-collagen antibody levels were also depressed in treated mice, consistent with the ability of Rac1 inhibitory peptide to suppress TCR/CD28 –induced TNFα, IFNγ, IL-17, ICOS and CD154 expression, gene products needed for follicular T cells to provide B cell help, in T cells from collagen-primed mice [146].
Much of our understanding as to how Rho family GTPases might contribute to pathology in RA stems from the finding that statins, hydroxymethylglutaryl-coenzyme A (HMG-CoA) inhibitors interfere with small GTPase function by depleting farnesyl pyrophosphate and geranylgeranyl pyrophosphate intermediates needed for localization and function of Ras and Rho family members, as well as G-coupled protein receptors and other signaling proteins [147]. In RA FLS, lipophilic statins (including simvastatin, fluvastain, pitavastatin, and cerivastatin) but not hydrophilic statins (such as atorvastatin) reduce proliferation and induce apoptosis, associated with inactivation of Rac1 and RhoA [148-150]. Simvastatin reduces membrane localization of both Rac1 and RhoA, and enhances both basal and TNFα-induced apoptosis, possibly due to decreased PKB activity [148]. Although the apoptotic PKB inhibitory effect of statins can be in part mimicked by silencing of Rac1, a more robust phenocopy is observed in RA FLS treated with an inhibitor of ROCK, a downstream effector of RhoA, or cell-permeable inhibitory peptides targeting RhoA [148, 149, 151]. Complementary use of RA FLS transfected with dominant-negative RhoA also demonstrates that RhoA is responsible for simvastatin interference with TNFα-induced NF-κB activation and IL-6 production [150]. Similar RhoA-dependent effects of simvastatin are observed with TLR2-stimulated peripheral blood monocytes from RA patients [152]. As in the case of Rac, few studies are available examining the effect of Rho inhibition in animal models of arthritis, but ROCK inhibitors reduce pain in rat models of arthritis [153]. Additionally, some studies have reported beneficial effects of statins in the treatment of murine CIA, although independent studies have failed to reproduce these results [154, 155].
Rho family signaling may also be important in SLE. In macrophages isolated from mice with spontaneous SLE-like disease, including MRL/lpr and New Zealand Black/White F1 (NZBWF1) mice, serum-dependent activation of RhoA, but not Rac, Cdc42 or Ras proteins is suppressed [156]. The genetic or molecular mechanism underlying this is currently unknown, and similar studies on SLE patient macrophages have yet to be reported. No studies are currently available in which Rho family GTPases were specifically targeted in animal models of SLE, but treatment of NZBWF1 mice with the statin atorvastatin decreases spontaneous lupus-like pathology, including diminuation of serum and glomerular auto-antibody levels [157].
FUTURE DIRECTIONS
As our understanding of the genetics, epigenetics, and biochemical signal cascades imprinting and driving the chronic inflammatory state in RA and other inflammatory diseases accelerates, it is clear that altered signaling of small GTPases contributes to the “semi-transformed” phenotype of inflamed tissue. However, much work remains to be done, in both understanding how specific GTPase signaling pathways contribute to disease, and identifying selective therapeutic strategies for modifying their function. We still lack a detailed overview of the local expression patterns of GTPase regulatory proteins at the site of inflammation of RA and other inflammatory diseases, and exceedingly few mice lacking small GTPases or their regulatory proteins have been examined in animal models of diseases. Such efforts are warranted by initial findings that inhibition of Rho signaling pathways, or activation of T cell Rap1 signaling, is protective in animal models of multiple sclerosis [158-161]. Another area requiring attention is that, in cases where genetically modified mice show altered adaptive immune phenotypes, it will be important to distinguish whether the phenotype arises from changes in lymphocyte function relevant to the human disease in question, or from changes in shaping of the antigen receptor repertoire during development [162-164].
An increasingly detailed understanding of how GEFs and GAPs are regulated and interact with their GTPase targets is providing new opportunities for therapeutic development (Fig. 3) [22]. To date there has been little attempt to directly target individual small GTPases either by interfering with their ability to bind to GTP, or by accelerating their instrinsic GTPases activity [22]. Some effort has been placed in exploring the possibilities of interfering with GTPase function using compounds which cause their mislocalization within the cell. However, in limited clinical trials, statins have demonstrated at best limited efficacy in the treatment of RA [147]. This may be because statins interfere with both the farnesylation and geranylgeranylation of Ras and Rho family GTPases. At least in RA FLS, these two families of small GTPases appear to antagonize each in terms of FLS MMP-1 production, leading to a neutral effect of statins on cellular activation [56]. FTIs are used in the treatment of cancer, while gerangeranyl transferase inhibitors are too toxic for use in animal models of arthritis [147]. The major caveat in further exploration of FTIs is that they will indiscriminately interfere with the function of not only Ras proteins, but also a subset of Rho family members and G-coupled protein receptors [55]. More specificity might be obtained using cell-permeable GTPase inhibitory fragments, but these compounds are cleared very quickly and are relatively unstable in vivo, limiting their efficacy.
One promising avenue of approach is the design of small compounds which specifically activate or inhibit GEFs and GAPs via interaction with allosteric regulatory domains or their catalytic sites. One example of the former is a cAMP-analog which specifically activates the Rap1 GEF EPAC, but not protein kinase A [165]. The generation of a series of inhibitors blocking DOCK2 catalytic activity provides proof-of-principal evidence that specific GEF activity can be targeted [129-131, 166]. Detailed structural analyses of small GTPases with their regulatory proteins and effectors is also allowing the development of biphasic aptamer compounds which block the interface of protein-protein interactions. Two such compounds have recently been identified, based on either chemical-genetic approaches, or chemical-based virtual screening technologies, which selectively interfere with activation of Rho GTPases by the GEF Trio [167, 168]. As detailed elsewhere in this journal series, signaling enzymes downstream of small GTPases, such as PI3-K, MAP kinases, and ROCK represent exciting targets for pharamacological intervention in IMIDs. Lastly, antibody strategies aimed at agonizing or antagonizing cell surface proteins coupled to the regulation of GEFs and GAPs is an area which has not been fully explored. Unintentionally, the clinical success of Abatacept (CTLA4-Ig) in the treatment of RA may already suggest the power of this technique, as one of the proximal effects of CTLA4-Ig on synovial T cells is the restoration of Rap1 signaling, an effect likely mediated by CD28 and CTLA4 functional interactions with RapGAPI [91, 98]. Time and effort will tell if biologicals represent an effective strategy for specifically interfering with the complexities and redundancies of small GTPases in chronic inflammatory diseases.
ACKNOWLEDGEMENTS
We would like to thank Aleksander M. Grabiec (our institute), for his asssistance in creating illustrations included in this manuscript.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.

