All published articles of this journal are available on ScienceDirect.

The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases
Abstract
The class 1 PI3Ks are lipid kinases with key roles in cell surface receptor-triggered signal transduction pathways. Two isoforms of the catalytic subunits, p110γ and p110δ, are enriched in leucocytes in which they promote activation, cellular growth, proliferation, differentiation and survival through the generation of the second messenger PIP3. Genetic inactivation or pharmaceutical inhibition of these PI3K isoforms in mice result in impaired immune responses and reduced susceptibility to autoimmune and inflammatory conditions. We review the PI3K signal transduction pathways and the effects of inhibition of p110γ and/or p110δ on innate and adaptive immunity. Focusing on rheumatoid arthritis and systemic lupus erythematosus we discuss the preclinical evidence and prospects for small molecule inhibitors of p110γ and/or p110δ in autoimmune disease.
OVERVIEW
The inflammatory response is a complex and highly regulated consequence of stimuli received from pathogens or damaged cells. Inflammation is triggered by bacterial derivatives (e.g. LPS or fMLP), molecules released from damaged cells (e.g. ATP) and chemical mediators (such as complement) which activate resident cells of the innate immune system to engulf pathogens and release pro-inflammatory cytokines to recruit leucocytes to the inflamed tissue. Subsequently, T and B lymphocytes participate in a targeted and specific response following presentation of pathogen-derived or disease associated antigens [1].
This powerful system is critical for defence against pathogens. The severe and often lethal phenotypes exhibited by patients with defects in immune cell function illustrate all too clearly the importance of immune system integrity. However this essential system has great potential to harm the host. Lymphocytes gain tolerance to host tissues through clonal deletion, anergy or via suppression by regulatory T cells. When these processes fail autoreactive lymphocytes can cause autoimmune and inflammatory conditions, often resulting in significant morbidity [2, 3].
The diverse roles served by PI3K in the innate and adaptive immune system offer hope that inhibition of this axis may provide new treatments for autoimmune conditions [4]. The ultimate goal must be to alleviate pathological inflammation while leaving resistance to infection intact. In this review we discuss the current understanding of class 1 PI3K signal transduction, with a particular focus on the PI3K isoforms p110γ and p110δ which are enriched in leucocytes. We examine how targeting these isoforms shows promise as a therapeutic strategy in autoimmunity, focussing on two systemic rheumatic diseases, rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), but also allude to other immune-related conditions that may be treated using PI3K inhibitors.
PHOSPHOINOSITIDE 3-KINASES
Phosphoinositide 3-kinases (PI3Ks) are lipid kinases that phosphorylate the 3-hydroxyl position of the inositol ring of phosphatidylinositol (PI) substrates within the plasma membrane and intracellular compartments. These substrates comprise PI, phosphatidylinositol-4-phosphate (PIP) and phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2). The phosphorylated effector products recruit and activate proteins expressing the lipid-binding Pleckstrin Homology (PH), phox homology and FYVE domains. PI3Ks control a wide variety of cellular processes, including migration, proliferation, differentiation and vesicular trafficking [5-7].
PI3Ks are classified according to their sequence homology and substrate specificity. Class I PI3Ks convert PI-4,5-P2 into PtdIns-3,4,5-triphosphate (PIP3), class II PI3Ks convert PI-4-P into PI-3,4-P2 and PI into PI-3-P, while the class III PI3K converts PI into PI-3-P. Class I PI3Ks are the most extensively studied and form the principle focus of this review. The biological functions of Class II PI3Ks are less well understood; the single class III PI3K enzyme Vacuolar Protein Sorting 34 (VPS34) is involved in vesicular intracellular trafficking and autophagy (Table 1).
Catalytic and Regulatory Subunits of PI3Ks and their Lipid Substrates and Products
| Catalytic Subunit | Regulatory Subunits | Substrate | Product | |
|---|---|---|---|---|
| Class 1A | p110α p110β p110δ |
p85α, p85β, p55α, p55γ, p50α | PI-4,5-P2 | PIP3 |
| Class 1B | p110γ | p101, p84 | PI-4,5-P2 | PIP3 |
| Class II | PIK3-C2α PIK3-C2β PIK3-C2γ |
PI-4-P PI |
PI-3,4-P2 PI-3-P |
|
| Class III | VPS34 | p150 | PI | PI-3-P |
Note that while there are three different types of PIP2 (PI-3,4-P2, PI-3,5-P2 and PI-4,5-P2), there is only one kind of PIP3 headgroup (PI-3,4,5-P3),
| Model | Details | Cells /Conditions Studied |
|---|---|---|
|
|
||
| PI3Kγ-/- [42-44] | PI3Kγ knock-out | T cells [43,47-50] |
| NK cells [24, 51-53] | ||
| Neutrophils [42-4, 54-57] | ||
| Mast cells [58] | ||
| Dendritic cells [59] | ||
|
|
||
| PI3Kγcx/cx [60] | Constitutively active PI3Kγ | Neutrophils and macrophages. |
|
|
||
| PI3KγKD/KD [12] | Kinase-inactive PI3Kγ | Neutrophils and macrophages. |
|
|
||
| PI3Kδ-/- [45, 46] | PI3Kδ knock-out | B cells [45, 46, 61-63] |
| T cells [45, 64] | ||
| NK cells [51, 53, 65] | ||
|
|
||
| PI3Kδ selective inhibitors [66] | IC87114 | B cells [67-69] |
| Neutrophils [56, 66, 70] | ||
| T cells [71-73] | ||
|
|
||
| PI3KδD910A/D910A [41] | Kinase-dead PI3Kδ | B cells [41, 67, 68, 74, 75] |
| T cells [41, 48, 72, 76-81] | ||
| Mast cells [82, 83] | ||
| Neutrophils [26, 56] | ||
| Dendritic cells [81, 84] | ||
| NK cells [24, 52] | ||
|
|
||
| PI3KγKO/δD910A [85] OR PI3Kγ-/-δ-/- [86, 87] |
PI3Kγ knock-out, PI3Kδ kinase-inactive; PI3Kγ and PI3Kδ knock-out.. |
T cells [85-88] |
| B cells [89] | ||
| NK cells [51] | ||
Murine Models of PI3K Activity in RA
| Model | Details | Isoforms Studied |
|---|---|---|
| CIA | Markedly reduced joint inflammation | Pharmacological p110γ or pan-PI3K inhibition [144, 146] |
| αCII-IA | Markedly reduced joint inflammation, reduced neutrophil invasion | Pharmacological and genetic p110γ inhibition [144] |
| Antigen-induced arthritis | Reduced joint inflammation, reduced activation and migration of phagocytes | Pharmacological and genetic p110γ inhibition[147] |
| K/BxN | Reduced joint inflammation, particularly in dual p110γ / p110δ inhibited mice, reduced LTB4 induced neutrophil accumulation | p110γ-/- p110δ-/- p110δ pharmacological inhibition Combined p110γ-/-p110δ-/-[119] |
Murine Models of PI3K Activity in SLE
| Model | Details | Isoforms Studied |
|---|---|---|
| Class 1A PI3K induced SLE (p65 regulatory subunit renders class 1A PI3K constitutively active, inducing SLE) | Prolonged lifespan, reduced CD4 cell survival, autoantibodies and glomerulonephritis | Genetic and pharmacological inhibition of p110γ(169) |
| MRLlpr | Prolonged lifespan and reduced glomerulonephritis, reduced lymphoid cells and autoantibodies | Pharmacological inhibition of p110γ(168) |
Other Autoimmune and Inflammatory Conditions with Preclinical Evidence for PI3K Inhibition as a Potential Treatment
| Model | Isoforms Investigated and References |
|---|---|
| Passive cutaneous anaphylaxis | p110δ [82, 83], p110γ [58] |
| Asthma/airway inflammation | p110δ [78, 147, 175-178], p110γ [127, 179], dual p110δ and p110γ inhibition [180] |
| Experimental autoimmune encephalitis | p110δ [102] and p110γ [109, 122] |
| Atherosclerosis | p110γ [181] |
| Contact and delayed type hypersensitivity | p110δ [71], p110γ [59] |
PI3Ks have diversified throughout evolution: yeast have only a class III PI3K enzyme, Caenorhabditis elegans and Drosophila melanogaster have single isoforms of each of the class I, II and III enzymes, while mice and humans have four class I isoforms, three class II isoforms and one class III PI3K [8].
The class 1A PI3K enzymes are heterodimers comprised of a regulatory subunit (p85) and a catalytic subunit (p110). Five regulatory Src Homology 2 (SH2) domain containing subunits have been identified; p85α, p50α, andp55α, (from alternative transcripts encoded by the Pik3r1 gene), p85β (encoded by Pik3r2), and p55γ (encoded by Pik3r3). Each of these regulatory subunits associates with any one of three catalytic subunits, p110α, p110β and p110δ. Class 1A PI3K regulatory subunits contain SH2 domains which enable recruitment to tyrosine phosphorylated proteins at the plasma membrane, thus receptor tyrosine kinases (RTKs) form their principle means of activation. The p110γ catalytic subunit associates with either p101 or p87/p84 to form the class 1B PI3K heterodimer which is generally activated by G-coupled protein receptors (GPCRs) [5-7].
p110α and p110β are ubiquitously expressed whereas p110γ and p110δ are enriched in leucocytes. p110δ is also expressed in some neurons [9] and cancer cell lines [10], but has low expression in other tissues. p110γ is expressed in cardiac myocytes and plays important roles in cardiac physiology, both as a kinase and as an adapter regulating cAMP-mediated signalling cascades [11-14].
The preferential signalling through p110γ and p110δ isoforms in leucocytes has prompted considerable pharmaceutical interest [4]. Targeted inhibition of these isoforms should provide immunotherapeutic benefit while minimising organ toxicity. Indeed, compounds inhibiting p110δ are already being assessed in clinical trials for the treatment of haematological malignancies [15-18]. Pan-PI3K and p110α-selective inhibitors are being evaluated in clinical trials for cancer [19].
ACTIVATION OF PI3KS BY TYROSINE KINASE AND/OR G PROTEIN COUPLED RECEPTORS
PI3K p110γ and p110δ signalling is initiated by different cell surface receptors in different cell types of the immune system, transmitting signals promoting growth, activation and differentiation. Key signalling cascades are initiated in neutrophils, macrophages, dendritic cells, T cells, B cells and mast cells (summarised in Fig. 1).

p110γ and p110δ are involved in the response to various cell signalling cascades that depend on cell type.
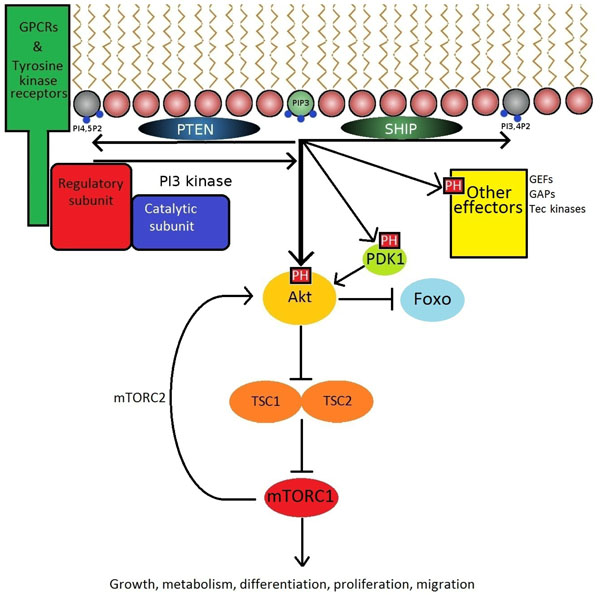
The production of PIP3 is a critical signalling hub. GPCR's and receptor tyrosine kinases expressing a YxxM motif recruit PI3K to the plasma membrane via interactions with SH2 domains. PI3 kinases then catalyse the formation of PIP3 from PI-4,5-P2. PIP3 then acts on a variety of downstream effectors expressing PH domains to promote cellular responses.
All four of the class 1 PI3K's have a Ras binding domain (RBD) in their catalytic subunit and the physiological role of Ras in PI3K activation has been elucidated by generating mice with mutations in the p110α or p110γ RBDs. In p110γ signalling, Ras has been shown to contribute to normal neutrophil function [20] whereas mice with a mutation in the p110α RBD show impaired lymphatic vessel development and reduced viability and resistance to Ras oncogene-induced tumourigenesis [21]. The Ras-related protein TC21 contributes to basal activation of p110δ in B cells and T cells and TC21-/- mice show some similarities with p110δ-deficient mice [22].
The simple concept that class 1A PI3Ks are activated by growth factor and cytokine RTKs and the class 1B PI3K is activated by chemokine responsive GCPRs does not always hold true. For instance, it is becoming increasingly clear that GCPRs can activate a wider range of PI3K isoforms than initially thought. p110δ can be activated by GPCRs in B cells [23] and in natural killer cells [24], and p110β can be activated by GPCRs in cells where p110γ is not highly expressed or when tyrosine kinase and GPCR signals merge [25, 26]. Conversely, the p84/p110γ heterodimer was shown to be activated by tyrosine-kinase linked receptors in macrophages by a Ras-dependent mechanism [27].
NEGATIVE REGULATION OF CLASS 1 PI3K SIGNALLING
The PI3K-Akt signalling axis has a profound influence on cellular functions, and must therefore be tightly regulated. Constitutive activation of class 1 PI3K signalling predisposes to autoimmunity and cancer, while inactivation of isoforms has varying effects ranging from embryonic lethality to immune deficiency, demonstrating the critical importance of maintaining physiological balance in this cellular cascade. The tight control of PIP3 levels within the plasma membrane is controlled by phosphatases including PTEN and SHIP which dephosphorylate PIP3 on the D3 and D5 positions, respectively. Deletion of SHIP and/or PTEN in lymphocytes leads to a variety of autoimmune syndromes and leukaemia [28-31]. Interestingly, drugs that activate SHIP may be used as anti-inflammatory drugs, presumably by inhibiting PI3K signalling in blood cells [32].
DOWNSTREAM SIGNALLING
The phosphorylation of PI-4,5-P2 to PIP3 at the inner leaflet of the cell membrane by PI3K permits the activation and recruitment of effector proteins expressing PH domains, including PDK1, Akt (also known as Protein Kinase B), guanine nucleotide exchange factors (GEF), and Tec family kinases. The best studied of these is the serine/threonine kinase Akt which can phosphorylate a large number of substrates [33]. Among these is the mammalian Target of Rapamycin mTOR [8] (Fig. 2). Rapamycin-based drugs are commonly used immunosuppressants in kidney transplant recipients [34]. Although rapamycin has been evaluated as a potential therapeutic in RA [35] and SLE [36], the associated adverse effects may be too severe for routine use in autoimmunity.
Insights from Mouse Models
The interrogation of signalling pathways in which the class I PI3Ks play a key role has been facilitated by the availability of small molecule inhibitors with high selectivity for the various class 1A isoforms and the development of mice with inactivating mutations in PI3K genes. These strategies provided a considerable advance on earlier approaches which utilised wortmannin or LY294002 (pan-PI3K inhibitors with known off-target effects on class II and III PI3K's, mTOR and casein kinase 2) allowing the dissection of the contributions of specific PI3K isoforms to lymphocyte signalling, trafficking, proliferation and development.
Homozygous deletion of the gene for p110α or p110β results in early embryonic lethality [37, 38]. Mice homozygous for an inactivating mutation in p110α also die as embryos [39], whereas mice with an inactivating mutation of p110β are born at reduced Mendelian ratios, yet survive into adulthood indicating that an unexpected non-catalytic function of p110β partially protects against this mortality [25, 26, 40].
By contrast mice with genetic inactivation of p110γ or p110δ are born at normal Mendelian ratios and live a normal life span despite having impaired immune cell responses [41-46]. The burgeoning interest in p110γ and p110δ as therapeutic targets in autoimmunity has been built on substantial data emerging from the study of mice with genetic and pharmacological inactivation of these isoforms, examples of which are detailed in Table 2.
PI3K IN B CELLS
The key role for PI3K in B cell physiology was first demonstrated by p85α-/- mice which showed impaired B cell development and humoral immune responses [93, 94], and this phenotype was also seen in mice with p110δ deletion or inactivation [41, 45, 46]. In particular, mice lacking p85α or p110δ have markedly reduced B1 cells (self-renewing IgM producing B cells populating the pleural and peritoneal cavities) and marginal zone B cells, with a lesser reduction in follicular B cells. In vivo, both the PI3K isoforms p110α and p110δ contribute to tonic survival signals generated by the B cell receptor (BCR) in absence of antigen, whereas p110δ is preferentially activated by acute BCR signalling [75]. Reduced homing to Peyer's patches and splenic white pulp cords is seen in adoptively transferred kinase-dead p110δ B cells, reflecting a role for p110δ in B cell chemotaxis [23]. Mice lacking p85α or p110δ exhibit reduced T-independent antibody responses and p110δ inactive B cells have reduced proliferative responses to anti-IgM, anti-CD40 and interleukin(IL)-4 stimulation [41, 46, 67]. Furthermore, TLR9-mediated antibody class switching is blocked as a result of p110δ inactivation [95]. However, p110δ signalling also negatively regulates class switch recombination through activation-induced cytidine deaminase (AID) expression during germinal centre responses such that inhibiting p110δ can increase class switching [68, 69, 96].
Although much is known about the central role played by p110δ in development and activity of conventional B cells, its precise function in the recently described regulatory B cell subset has not been reported. Regulatory B cells were first identified in mice and subsequently in humans, and mediate their regulatory function via the production of IL-10 [97-99], leading some authors to describe them as B10 cells [96]. In humans, IL-10 producers comprise 1-5% of circulating B cells, although cells with the potential to produce IL-10 may be found at higher frequencies. Abnormalities in the number [95] or function [99] of regulatory B cells have been described in patients with autoimmune diseases, suggesting that this subset may be of relevance to the development of treatments for autoimmunity.
Consistent with an important role for PI3K in B cell homeostasis, patients with B cell malignancies have responded well to the p110δ-selective inhibitor CAL-101. In one study, all chronic lymphocytic leukaemia (CLL) patients treated with this inhibitor showed a reduction in lymph node size which was often associated with an increase in blood lymphoblast count [18]. However, patients who stopped taking the drug showed recurrent lymph node enlargement suggesting that the drug had not necessarily just killed the leukaemia cells - rather, these results raise the possibility that p110δ is required for stromal cell support of proliferating CLL cells, perhaps by virtue of its role in chemokine receptor signalling. In vitro studies of the effect of CAL-101 on CLL cell viability showed that it was able to block the pro-survival effect of BCR cross-linking with IgM, as well as reducing elevated CCL3 and CCL4 levels [15, 100]. Further studies have shown that CAL-101 inhibits CLL cell chemotaxis and migration beneath stromal cells and sensitises CLL cells toward standard chemotherapeutic agents [101].
PI3K IN T CELLS
p110δ-deficient mice have relatively normal numbers of thymocytes, while p110γ-deficient mice have a modest reduction in numbers [43]. However, combined p110γ/ p110δ deficiency has a much more significant impact on thymocyte numbers, with substantially reduced transition through the CD4-CD8- stage and reduced survival of cells at the CD4+CD8+ stage [86, 87]. The apparent redundancy of p110δ and p110γ during T cell development is surprising, given that these PI3K isoforms are activated by distinct receptors. This conundrum was recently resolved by demonstrating a requirement for CXCR4 signalling in the thymus which was mostly dependent on p110γ, whereas pre-TCR signalling was entirely dependent on p110δ [88]. Elevated Th2 cytokines and eosinophilia observed in p110δ-p110γ double deficient mice may well be a consequence of impaired T cell development and subsequent lymphopenia induced proliferation of the few T cell that do develop [85].
T helper cell differentiation and cytokine production is PI3K dependent in mice and humans as demonstrated by the ability of p110δ-selective inhibitors to block IFN-γ, IL-4 and IL-17 indicating that PI3K function is required in differentiated T helper cells [71]. Akt and Foxo signalling downstream of the TCR and CD28 is attenuated in p110δD910A mice with impaired differentiation along the Th1 and Th2 lineages [76] and p110δD910A mice exhibited significantly decreased Th2 cytokine responses and were protected against eosinophilic airway inflammation [78]. Moreover, p110δD910A mice were resistant to experimental autoimmune encephalitis which was correlated with reduced numbers of Th17 cells [102].
PI3K also plays an important role in T follicular helper (TFH) cells, a subset of T cells which are critical for the provision of help to germinal centre B cells during affinity maturation and for their differentiation into long-lived plasma cells [103]. TFH cells are likely to be crucial pathogenic mediators in SLE, as it is the long-lived plasma cells they create that exhibit most resistance to B cell depleting therapies and are the predominant source of high-affinity pathogenic autoantibodies [104, 105]. The ability of the costimulatory ICOS to induce TFH cells and to promote the generation of germinal centres and high affinity, class switched antibodies is critically dependent on p110δ [64, 106].
Regulatory T cells (Tregs) develop both in the thymus and the periphery from mature CD4 T cells and serve a critical role in controlling the adaptive immune system and preventing unrestrained inflammatory responses [107]. p110δD910A mice has revealed increased numbers of Tregs in the thymus, but reduced numbers in the spleen and lymph nodes implicating a role for p110δ in the maintenance of Tregs in the periphery [79, 108]. p110δ-inactive Tregs also fail to suppress T cell proliferation in vitro, to secrete the key suppressive cytokine IL-10 and to prevent a model of inflammatory bowel disease [79]. Interestingly, p110δ-deficient mice show enhanced clearance of the protozoan parasite Leishmania major, despite a major reduction in Th1 cells [81], in part as a consequence of altered Treg function. mTOR deficient T cells display normal activation and IL-2 production on stimulation, but impaired differentiation into Th1, Th2 and Th17 cell subsets; instead T cells differentiated into Foxp3+ regulatory T cells [2, 109, 110]. Rapamycin also promotes the expansion of Tregs [111]. Moreover, Foxo transcription factors control the pivotal transcription factor Foxp3 which is both necessary and sufficient for the induction of regulatory T cell (Treg) function [112]. PI3K signalling can therefore suppress Foxp3 expression by activating Akt which phosphorylates and sequesters Foxo1 and Foxo3a [1]. The ability of PI3K to activate mTOR and inhibit Foxo may explain the increased abundance of Tregs in the thymus, but does not explain the paucity of Tregs in the spleen of p110δD910A mice [79, 108].
PI3K IN NEUTROPHILS
Neutrophils are the most abundant circulating phagocyte and form the cornerstone of the innate immune system, playing a central role in the destruction of invading pathogens [113]. Neutrophil dysfunction is also known to contribute to the pathogenesis of a number of autoimmune diseases, including SLE and RA. Thus, inhibition of neutrophil activation is a potential therapeutic goal in the treatment of these conditions and pathways implicated in dysfunctional intracellular signalling may provide promising routes of enquiry.
In common with other leucocytes, the p110γ and p110δ isoforms of PI3K are markedly enriched in neutrophils. However, although both p110γ and p110δ are both expres-sed in neutrophils, the upstream RTKs and GPCRs that recruit and activate them are downstream of a markedly different array of cell surface receptors to those implicated in PI3K signalling in lymphocytes. Integrins, Fcγ receptors and cytokine receptors are key among RTK stimuli, and receptors for fMLP, C5a and LTB4 are all known to activate GPCR-mediated PI3K activity [114].
Effective neutrophil function is dependent on several related activities ranging from chemotaxis, endothelial adhesion, diapedesis, phagocytosis, activation, phagolysosomal maturation, free radical production and (later) apoptosis. Class 1 PI3K control neutrophil spreading on endothelium [115], migration [57, 70] and pathogen killing [116]. In addition the class 3 PI3K VPS34 is important in phagocytosis [117].
PI3Kγ genetic knockout in mice results in fully differentiated neutrophils that fail to phosphorylate Akt and show impaired respiratory burst, motility and migration toward chemotactic stimuli, especially in vivo(42). PI3K activity through the p110γ isoform can contribute to cell polarisation, with PIP3 generation at the leading edge involved in actin rearrangement [54, 118]. However an important role also exists for SHIP, with SHIP-/- neutrophils demonstrating impaired localisation of Akt at the leading edge of the plasma membrane reducing motility [108].
In vitro the impairment of neutrophil directional movement by p110γ inactivation is exacerbated by pharmacological inhibition with the p110δ-specific inhibitor IC87114 [66]. A phenotype reproduced in in vivo models of pulmonary inflammation and arthritis [119]. The effects on neutrophil migration are mediated through both reduced neutrophil tethering in TNF-α primed blood vessels (p110γ-mediated) and via impaired transmigration of neutrophils across vessel walls (p110δ-mediated) [120].
At the site of infection, neutrophils kill pathogens by generating reactive oxygen species (ROS) via the respiratory burst process. Genetic inactivation of p110γ impaired ROS production in neutrophils in response to fMLP(42), in keeping with a likely impairment in killing of phagocytosed pathogens. p110δ is also implicated in neutrophil respiratory burst in human (but not mouse) neutrophils stimulated with fMLP, where initial transient p110γ-dependent PIP3 generation is followed by more sustained p110δ-mediated PIP3 production [56].
Neutrophil PI3K signalling is not wholly dependent on p110γ and p110δ as the p110β isoform has also been shown to play a critical role in FcγR-dependent activation of mouse neutrophils by IgG immune complexes. ROS production in response to low concentrations of immune complexes is completely blocked by p110β inhibition. At higher concentrations of immune complexes, combined inhibition of p110β and p110δ was required to achieve the same suppression of ROS. In an in vivo model of FcγR-dependent inflammatory arthritis, combined p110β and p110δ deficiency provided almost complete protection from pathology [26].
The diverse and complementary influences of the various isoforms of PI3K on neutrophils suggest that drugs targeting p110γ and p110δ may exert immunosuppressive effects that could be of value in conditions in which neutrophil activation play a prominent role, and that strategies which both target single isoforms and combination inhibition may offer therapeutic benefit. Accompanying this potential is the concern that PI3K inhibition could increase susceptibility to opportunistic infections as suggested by the increased susceptibility to Streptococcus pneumoniae pneumonia seen in p110γ-/- mice [121]
PI3K IN OTHER CELLS OF THE IMMUNE SYSTEM
Mast cells reside in tissues such as the skin and mucosa and are characterised by the ability to release large amounts of heparin and histamine from their cytoplasmic granules. This release is mediated by the RTKs c-Kit and FcεR1 which couple to the p110δ isoform of PI3K. Mast cell degranulation plays an important role in allergic inflammatory disorders such as asthma and allergic rhinitis, and in anaphylaxis. Mice with inactive p110δ and those treated with a pharmacological p110δ inhibitor are protected from passive cutaneous anaphylaxis [82, 83]. p110γ inhibition and p110γ-/- mice have normal early mast cell responses but a reduced phenotype at later stages, suggesting an impairment in signal amplification [58].
Dendritic cells (DCs) are potent antigen-presenting cells with roles in initiating cellular and humoral immune responses and in tolerance induction. Prior to antigen encounter DCs reside in tissues in an immature state, becoming activated upon contact with antigen in the context of inflammatory or pathogen-derived stimuli. This activation leads to DC migration to the T-cell rich paracortex of draining lymph nodes where they present antigen to CD4 T cells, initiating immune responses. DCs have been studied in p110γ-/- mice, in which they demonstrate normal differentiation but have markedly impaired migration. This results in a reduction in severity of contact hypersensitivity in p110γ-/- mice [59] and may delay symptoms in an experimental autoimmune encephalitis [122]. The role of PI3K is not limited to p110γ; p110δ has a key role in promoting IL-6 release in response to cKit stimulation on dendritic cells by cholera toxin [84].
Macrophages coordinate C5a and FcγR signalling through the p110γ and p110δ isoforms of PI3K. In a mouse model of the passive reverse lung Arthus reaction, genetic deletion of p110γ blocked C5aR signalling crucial for activation of lung macrophages. However C5a production occurred normally in p110γ-/- mice but was impaired in p110δ-/- mice, which demonstrated resistance to acute immune complex-induced lung injury, thus defining p110δ as a crucial element of FcγR signalling in the production of C5a [123].
Eosinophils infiltrate release pro-inflammatory cytokines and ROS in response to IgE immune complexes. Studies using the pan-PI3K inhibitor wortmannin have implicated PI3K in eosinophil chemotaxis and release from the bone marrow [124], and inhibitor-treated mice show a reduction in eosinophilia in allergen-induced bronchial inflammation [125]. Subsequent studies suggest that p110γ is the isoform most likely mediating this effect [126, 127]. However, some data suggest that combined PI3K inhibition may also have unwanted effects on eosinophils as p110δ-p110γ double-mutant mice developed marked eosinophilic infiltration in multiple organs, with increased serum IgE, IL-4 and IL-5 [85].
PERTURBATIONS OF THE PI3K PATHWAY IN RHEUMATIC DISEASES
Rheumatoid Arthritis
RA is characterised by synovitis and systemic inflammation secondary to autoantibody production (particularly rheumatoid factor and citrullinated peptide). It is common, disabling and can have a significant impact on patients' quality of life [128]. Current treatment strategies centre on the use of disease-modifying antirheumatic drugs (DMARDs) as a first line treatment [129-132]. However, these are agents are non-specific, may be associated with problematic side effects and disease flares occur despite such treatments. Advances in RA therapies have been considerable in recent years with a number of biological agents receiving approval. These include TNF inhibitors [133], a CTLA-4 fusion protein (abatacept) [134], a B cell depleting CD20 antibody (rituximab) [135] and an IL-6 inhibitor (tocilizumab) [136]. Although these biological treatments have demonstrated impressive efficacy in subsets of patients, their widespread use remains limited by cost and susceptibility to infection [137]. Furthermore, not all patients respond to such treatments. Thus there remains an unmet need to develop further targeted therapies that may benefit patients with RA.
The underlying pathogenesis of RA is complex and can involve inappropriate activation of cells of both the innate and adaptive immune systems with T cells [138], B cells [139], dendritic cells [140], mast cells [141] and neutrophils [142] all implicated in disease models and human pathology.
RA is thought to be initiated in peripheral lymphoid organs by the presentation of self-antigens to autoreactive T cells. This in turn prompts activation of autoreactive B cells to produce autoantibodies that form immune complexes. These deposit in joints triggering the release of pro-inflammatory cytokines from neutrophils, macrophages and mast cells that cause inflammatory cell migration and pannus formation. Tissue damage then results from the activation of synoviocytes which invade cartilage and osteoclasts which increase bone resorption [143]. Over time irreversible bone and cartilage destruction occurs, leading to accrual of disability [128].
The p110γ and p110δ isoforms of PI3K have attracted considerable interest as pharmacological targets in the treatment of RA given the known effects of their inhibition on a variety of immune cells [4] (Table 3). PI3Kγ blockade by both genetic and pharmacological approaches suppresses joint inflammation and damage in two murine models of RA - collagen-induced arthritis (CIA) and αCII-induced arthritis (αCII-IA). In the CIA model mice treated with AS-605240 (a p110γ-selective inhibitor) showed a significant reduction in joint inflammation with a near normal mean paw thickness. There was also marked amelioration of histological measures of synovial inflammation, cartilage erosion and neutrophil infiltration in arthritic joints. Similar findings were observed in the αCII-IA model using both mice with genetically inactive p110γ and pharmacological inhibition with the same small molecule inhibitor. Similarly p110γ-deficient and p110γ inhibitor treatment ameliorated disease in the early stages of antigen-induced arthritis [144]. A further role for p110γ is seen in the regulation of synovial fibroblasts in RA, in which p110γ deficiency leads to a milder inflammatory erosive arthritis and TNF-α mediated cartilage destruction. p110γ is highly expressed in the synovium of patients with RA [145].
The role of p110δ in RA has also been investigated. In a study utilising the neutrophil-dependent K/BxN serum transfer model of arthritis, genetic and pharmacological inactivation of p110δ reduced joint injury. Interestingly the effect of pharmacological inhibition of p110δ in mice with genetic inactivation of p110γ was also assessed and was found to totally prevent any macroscopic evidence of joint inflammation or histological evidence of bone or joint damage. Consistent with this a marked reduction in neutrophil infiltration was observed, raising the possibility that combined p110γ and p110δ inhibition may potentially offer additive therapeutic benefits [119]. In a separate study, the pan-specific PI3K inhibitor ZST474 was shown to alleviate CIA by a mechanism that the authors suggested also involved the inhibition of osteoclast formation [146].
Given the 70 million years of divergent evolution separating rodents and Homo sapiens, murine models can only ever reflect human disease imperfectly. Although these models serve a vital role in supporting the development of isoform specific inhibitors for human use, certain pathophysiological differences between these models and human disease may be pertinent to PI3K signalling in particular. The CIA model, considered the most faithful model of human disease, shares many similarities with rheumatoid arthritis – an association with certain MHC Class II haplotypes, a breach of tolerance with generation of autoantibodies, and comparable clinical features. Yet the studies appraising p110γ inhibition focussed on the effect of reduced neutrophil and monocyte chemotaxis. In humans there is evidence to suggest that retention of inflammatory cells within the rheumatoid joint is a more critical factor than the rate of synovial infiltration [148]. Furthermore neutrophil accumulation is a prominent feature in CIA whereas in the rheumatoid synovium macrophages comprise the major phagocytic population. Additionally, collagen autoantibodies are critical players in CIA pathology whilst human RA patients do not form autoantibodies against this antigen, and it is not certain that B and T cell antigen receptor signalling is actively engaged in established rheumatoid arthritis. Similar drawbacks exist with the K/BxN and serum transfer models of arthritis which again depend on non-RA autoantibodies and neutrophil infiltration [149, 150]. Nevertheless, murine models have been invaluable in supporting the progress of many recent drugs to the clinic and will continue to serve an essential complementary role to human ex vivo studies in the preclinical arena.
In vitro studies on human cells using p110δ inhibition showed that T cells from healthy volunteers stimulated with anti-CD3 and anti-CD28 had reduced Akt phosphorylation and IL-17, TNF-α and IFN-γ production. Similar results were found in mononuclear cells isolated from the synovial fluid of patients with reactive arthritis, with IFN-γ, TNF-( and IL-17 production blocked to a similar extent to that observed with ciclosporin [71]. The effect of inhibiting PI3K with the broad spectrum inhibitors LY294002 and wortmannin has also been evaluated. PI3K was shown to be necessary for TGF-β mediated proliferation and resistance to Fas-mediated apoptosis in patient synovial fibroblasts [151] and for the ability of anti-CD3-stimulated RA patient synovial T cells to induce monocyte TNF-α production [152]. Further interest in PI3K signalling was stimulated by the observation that RA patient synovial fibroblasts have markedly reduced expression of PTEN, in keeping with over activity of signalling in this axis [153]. In addition, macrophages have PI3K-dependent expression of the pro-survival protein Mcl-1, and LY294002 induces apoptotic death in RA patient synovial macrophages [154]. PI3K signalling is also necessary for T cell-induced IL-10 (an inhibitory cytokine) production by macrophages [155].
Given that drugs that target innate and adaptive immune responses have both shown efficacy in the clinic, the rationale for developing dual specific p110δ-p110γ inhibition in RA seems strong [156]. Studies that investigate the effect of isoform-specific inhibitors (both p110γ and p110δ individually and together) on patient-derived samples are needed to combine the encouraging findings from murine models with the known effects of pan-PI3K inhibition seen in earlier studies on patient samples.
SYSTEMIC LUPUS ERYTHEMATOSUS
SLE is a clinically and genetically heterogeneous autoimmune condition that displays a striking gender disparity, with females about nine times more likely to be affected than males. Common features include inflammatory arthritis, glomerulonephritis and a photosensitive rash, although numerous other tissues may be involved including the brain, lungs, heart and serosa. Significant advances in treatment have been achieved despite a 50 year period in which no new drugs were licensed for SLE. In the 1950s there was a 50% survival at 4 years, compared to an 80% 15 year survival rate today [157]. However the morbidity associated with SLE remains substantial, with patients suffering recurrent disease flares necessitating potent immunosuppression to achieve remission. As a result, a patient diagnosed with SLE at the age of 20 years still has a 1 in 6 chance of dying by the age of 35 [158].
The central pathogenic process in SLE is a loss of tolerance to nuclear antigens [159] such as chromatin, ribonucleoproteins (RNP's) and DNA. This results in the production of autoantibodies, that subsequently give rise to immune complex deposition in target organs. These deposited immune complexes initiate complement activation and cross-link Fc receptors on tissue macrophages and plasmacytoid dendritic cells, perpetuating the inflammatory response. An important component of this inflammatory response is IFN-α which promotes autoreactive T cell activation and B cell maturation and differentiation, resulting in self-perpetuating disease progression. Further actions of IFN-( include activation of neutrophils with the release of neutrophil extracellular traps (NETs), a process designed to entrap pathogens and facilitate their killing. In SLE this process further exacerbates IFN-α production driving disease flares and inflammation.
Current treatments are based on broadly acting immunosuppressants such as corticosteroids, azathioprine, mycophenolate and methotrexate but these drugs may be associated with problematic side effects. The recent approval by the FDA of the anti-BAFF monoclonal antibody belimumab for the treatment of SLE represents the first and only new treatment and has proven informative - not only in demonstrating the effectiveness of B cell targeted therapies but also in how to best design clinical trials for this condition characterised by high phenotypic heterogeneity [160, 161]. Initially belimumab failed at phase II to meet its co-primary endpoints of reduction in Safety of Estrogens in LupusErythematosus National Assessment–Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) score at week 24 and prolongation of time to first flare at 52 weeks. Belimumab's eventual success depended on the identification of a subset of patients with seropositive SLE that demonstrated reduced and stabilised disease activity, prompting the continuation of this study [162]. At phase III seropositive SLE patients randomised to belimumab demonstrated significantly higher SLE Responder Index (SRI) rates compared to placebo controls at 52 weeks and more had reductions of a least four points in their SELENA-SLEDAI scores at the same time point [160].
B cell activating factor belonging to the TNF family (BAFF) signalling is impaired in p110δ-deficient B cells, resulting in reduced survival, growth and proliferation in vitro [63]. p110δ has been found to be associated with the BAFF receptor, although another report suggested that the ability of BAFF to activate the PI3K pathway was indirectly mediated by an NF-κB target gene [163, 164]. Regardless of the precise mechanism involved, these data raise the possibility that one mechanism of p110δ inhibitors may be to attenuate BAFF-dependent pathology in SLE.
Several genetically engineered mouse strains with enhanced PI3K/Akt/mTOR signalling have been noted to develop a lymphoproliferative syndrome with autoimmune features resembling SLE (Table 4) [28-30, 165-167]. Conversely, promising results have been obtained from attempts to inhibit PI3K activity in mouse models of SLE [168, 169]. Inhibition of p110γ in the SLE-prone MRLlpr strain with the inhibitor AS605240 attenuated glomerulonephritis and extended lifespan, with 88% of treated mice surviving five months compared with only 60% of controls. Treated mice also had marked reductions in splenocyte numbers, lower DNA-specific autoantibody levels, less severe histopathological features of glomerulonephritis and a reduction in pathogenic CD4+ memory T cells [168].
In parallel experiments, the same authors used a second model of SLE caused by constitutive activation class 1A PI3K, and crossed these mice with p110γ deficient mice to assess the ability of p110γ to ameliorate lupus-like disease [169]. In this experimental system p110γ-deficient mice showed significantly prolonged survival with 70% still healthy at 16 months of age, compared with only 20% of SLE-prone controls, although T cell invasion and lymphoproliferation were unimpaired.
The role of p110δ-mediated PI3K signalling in SLE has not yet been directly studied in mouse models but is a promising area for future investigation. In San Roque mice, a lupus like syndrome results from the expansion of TFH cells secondary to elevated expression of the costimulatory receptor ICOS [170, 171]. In humans, an increased number of circulating TFH cells has been observed in a subset of patients with SLE with more severe pathology [172]. p110δ is essential for ICOS signalling and p110δ-deficiency severely impairs the development of TFH cells [64, 106]. Thus p110δ inhibitors may provide a means of blocking the development or maintenance of disease-associated TFH cells in patients with lupus.
PI3K activity was found to be higher in T cells from SLE patients than from normal controls and this increased PI3K activity appeared to be provided by elevated p110δ activity [173]. Interestingly, the PI3K activity was higher in T cells from patients SLEDAI scores of more than 4. The authors further provided evidence that the increased PI3K activity rendered the SLE T cells resistant to activation induced cell death. In a study of lipid uptake in SLE patient macrophages, scavenger receptor up-regulation in response to IFN-α was blocked by inhibition of the PI3K/Akt pathway with the pan-PI3K inhibitor LY294002 suggesting that PI3K signalling may contribute to foam cell formation. This could implicate a role for PI3K in the excess cardiovascular morbidity suffered by patients with SLE [174].
In summary, given the central role played by PI3Ks in T cell-dependent humoral immune responses and B cell homeostasis as well as promising pre-clinical studies suggests a strong rationale for the development of PI3K inhibitors for SLE.
OTHER AUTOIMMUNE AND INFLAMMATORY DISEASES
PI3K inhibitors have potential as treatments for a wide range of autoimmune and inflammatory conditions. Reviewing the preclinical evidence for all of these in details is beyond the scope of this article, but we have included a list of animal models in which p110γ and p110δ inhibitors or genetic knock-outs and knock-ins have shown efficacy in pre-clinical models in the table below (Table 5).
Translating the Bench-Top Science into Bedside Treatments
Selecting drugs to make the transition from preclinical models of diseases to effective therapies for human diseases remains a significant challenge. There is a substantial unmet need for new drugs to treat patients with RA resistant to anti-TNF based therapies, and SLE patients intolerant of (or poorly responsive to) current, commonly used immunosuppressants.
The promising results from CAL-101 (the first in class p110δ inhibitor) in its trials for refractory CLL and indolent non-Hodgkin's lymphoma provides some reassurance about the safety profile of drugs in this family [16, 17]. p110δ inhibitors are also being developed for other indications, including rheumatic diseases described in this review. Indeed a striking increase in patent applications for inhibitors claiming p110δ selectivity has occurred over the last few years [182].
Ongoing challenges facing the academic and pharmaceutical industries in the advancement of PI3K inhibitors include difficulties in synthesising highly specific p110γ inhibitors [156, 183]. The availability of crystal structures of p110δ and p110γ in complex with different inhibitors will no doubt accelerate progress in developing ever more selective and potent PI3K inhibitors [21, 112]. Whether dual inhibitors of p110δ and p110γ offer advantages over single isoform-selective inhibitors used alone or in combination will need to be explored further [156].
The wealth of preclinical evidence for p110γ and p110δ inhibition in autoimmune and inflammatory conditions suggests significant potential for patient benefit. The results of early clinical trials are anticipated with great interest as the roles of these immunologically important kinases become more clearly defined in humans.
ACKNOWLEDGEMENTS
We would like to thank Dalya Soond for constructive comments on the manuscript.
EBH is supported by the Wellcome Trust Translational Medicine and Therapeutics (TMAT) which is also funded by GlaxoSmithKline. Research in the Okkenhaug laboratory is supported by the BBSRC, Wellcome Trust and GlaxoSmithKline. KO is a consultant for GlaxoSmithKline.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ADDENDUM
Since this manuscript was accepted for publication key papers have shown that p110δ is a key regulator of synoviocytes function in rheumatoid arthritis. Using qPCR and immunohistochemistry the authors showed that p110δ mRNA and protein expression levels were higher in the synovial lining of patients with rheumatoid arthritis compared to controls with osteoarthritis. They went on to demonstrate that select pharmacological inhibition of p110δ could block PGDF and TNF-induced Akt activation [184]. Separately, the role of p110δ in autoimmunity has been investigated using a Lyn-deficient mouse. These mice develop autoreactive antibodies, glomerulonephritis and generalised inflammation secondary to a hyperactivated B cell phenotype with a phenotype that has certain similarities to human SLE. Mice heterozygous for the p110δD910A mutation had significantly attenuated disease parameters. Interestingly this protection was mediated in part as a consequence of greatly diminished T cell activation, despite the fact that Lyn is not expressed in these cells – implying an impairment of p110δ activity restricts the ability of T cells to induce B cell class switching and contribute to disease [185].

