All published articles of this journal are available on ScienceDirect.

The Role of TGF-β Receptors in Fibrosis
Abstract
Recent advances in defining TGF-β signaling pathways have provided a new level of understanding of the role of this pleiotropic growth factor in the development of fibrosis. Here, we review selected topics related to the profibrotic role of TGF-β . We will discuss new insights into the mechanisms of ligand activation and the contribution of Erk1/2 MAPK, PI3K/FAK, and Endoglin/Smad1 signaling pathways to the process of fibrosis. There is growing evidence of the disease-specific alterations of the downstream components of the TGF-β signaling pathway that may be explored for the future therapeutic interventions.
INTRODUCTION
Transforming Growth factor beta proteins (TGF-βs) are a group of three structurally similar proteins, TGF-β1, TGF-β2 and TGF-β3, that are the founding members of a larger family of proteins that comprise of BMPs (bone morphogenetic proteins), GDFs (growth and differentiation factors), activins, inhibins, and MIF (Müllerian inhibitory factor) [1]. TGF-β is an evolutionarily conserved multifunctional cytokine from invertebrates to human, with a diverse spectrum of biological activities ranging from the development of an embryo to the maintenance of tissue homeostasis [2]. Adding to the complexity, the functions of TGF-β are tissue specific and widely vary in a spatio-temporal manner. While a key role of TGF-β signaling in organ fibrosis is supported by ample evidence, specific mechanisms involved in deregulation of this pathway towards fibrotic outcomes vary depending on the affected organ. This review will highlight recent developments in this field focusing on the aspects of TGF-β signaling with the relevance to the process of dermal fibrosis.
ACTIVATION OF LATENT TGF-β
During physiological tissue remodeling expression and activation of TGF-β is tightly controlled, however deregulation of these processes is frequently associated with human fibrotic diseases. While many fibrotic disorders are characterized by the elevated levels of TGF-β, additional mechanisms could also account for the increased TGF-β signaling in fibrosis. High levels of TGF-β were observed in renal fibrosis where it was shown that all three TGF-β isoforms were overexpressed in glomerular and tubulointerstitial compartments of patients with glomerular diseases characterized by excessive extracellular matrix (ECM) deposition [3]. In addition, circulating and urinary levels of TGF-β were also highly elevated in patients with type II diabetes [4]. Elevated levels of TGF-β1 mRNA were found in fibrotic human lungs [5] and in BAL (bronchoalveolar lavage) fluids from patients with scleroderma (SSc, systemic sclerosis) [6]. Likewise, increased circulating levels of TGF-β1 were demonstrated in patients with hypertrophic and restrictive cardiomyopathy [7]. Interestingly, in SSc TGF-β mRNA was only detected in association with inflammatory cells, but not in sclerotic skin, underscoring an important role of elevated TGF-β in initiation, but not in progression of dermal fibrosis in SSc [8]. Furthermore, circulating active TGF-β levels were decreased in SSc and correlated inversely with the degree of skin fibrosis [9].
As TGF-β is secreted in inactive form, factors that contribute to its activation locally in the affected tissues might play a critical role in perpetuation of the fibrotic process during chronic stages of the disease. TGF-β is secreted and deposited into ECM as a large latent complex (LLC) that consists of latent TGF-β binding protein (LTBP) covalently bound to so called small latency complex (SLC). SLC is formed by a homodimer of TGF-β non-covalently bound to an RGD-containing N-terminal latency associated binding peptide (LAP) [10]. Recent studies provided compelling evidence for the integrin-mediated activation of latent TGF-β [reviewed in [11-13]]. LAPs of TGF-β1 and -3, but not TGF-β2, contain RGD motif and can be activated through this mechanism [13]. To date, six integrins, including all five αv integrins, as well as α8β1 have been shown to interact with the RGD-containing LAPs [13]. Integrins αvβ5 and αvβ6 were shown to activate TGF-β via a mechanism that does not require cleavage of latent TGF-β and most likely involves conformational change of the latent complex. This protease-independent mechanism entails contractile cell forces and can be enhanced by agents that stimulate cell contraction such as thrombin, angiotensin-II, endothelin-1 [14], as well as LPA [15]. A protease-dependent mechanism that does not involve cell contraction has been described for integrin αvβ8, which upon binding to SLC induces MT1-MMP (MMP14)-mediated release of TGF-β [16]. Other, non integrin, mediated mechanisms involve proteolytic cleavage of LAP by MMP2, MMP9 and plasmin [rev. in [17]]. Another proposed mechanism involves conformational rearrangement of the LAP-TGF-β complex upon interaction with thrombospondin-1 (TSP-1) [18] or a recently described ADAMTS1 (ADAM metallopeptidase with thrombospondin type motif, 1) [19]. Notably, there is evidence for the excessive activation of TGF-β in human fibrotic diseases. For example, SSc fibroblasts express elevated levels of integrin αvβ5 and αvβ3, which contributes to the activation of the autocrine TGF-β signaling and increased collagen synthesis in these cells [20, 21]. In addition, SSc fibroblasts produce more TSP-1, which may further augment autocrine TGF-β signaling [22, 23]. Importantly, expression of TSP-1 mRNA in the skin in vivo was highly correlated with degree of skin fibrosis in SSc patients [24]. The importance of αvβ6 integrin was demonstrated in animal models of fibrosis, including pulmonary and hepatic fibrosis [25]. Recently, elevated levels of ADAMTS1 were observed in human fibrotic livers. Additional functional studies demonstrated a key role of ADAMTS1 in activation of TGF-β signaling during experimental liver fibrosis [26]. Together, these clinical and experimental data support the view that deregulated activation of latent TGF-β plays an important role in the development of fibrosis.
TGF-β RECEPTORS - OVERVIEW
TGF-β receptors are transmembrane proteins with intrinsic serine/threonine kinases activity and include Type I (TβRI, also termed Activin Like Kinase 5, ALK5) and Type II (TβRII) receptors [27]. Both types of receptor have a short extracellular region, a transmembrane region and a large intracellular cytoplasmic domain. The extracellular domain undergoes glycosylation, and while the TβRII has a high affinity for the ligand, TβRI does not bind to TGF-β. The transmembrane domain of TβRII is constitutively phosphorylated at Ser213 independent of ligand activation and is essential for downstream signaling. In contrast, transmembrane region of TβRI is phosphorylated at Ser165 by TβR-II in a ligand dependent manner. Both TGF-β receptors have the intracellular domain with inherent serine/threonine kinase activity. In TβRI a unique glycine-serine region termed the GS domain is present between kinase and transmembrane domains [28]. Upon ligand binding, TβR-II recruits and activates TβRI by phosphorylating the GS domain. In addition to the major Type I and II receptors, accessory TGF-β receptor such as betaglycan and endoglin are present and collectively termed as Type III receptors. The major function of these co-receptors appears to be to increase the bioavailability of TGF-β to the signaling TGF-β receptors. Interestingly, recent studies have challenged previously held view that TGF-β receptors are present on the cell membrane as preformed homodimers. Zhang et al. have elegantly demonstrated that in the absence of TGF-β receptor isoforms are monomeric and dimerization takes place upon TGF- binding to TβRII [29]. Furthermore, Huang et al. have shown that TGF-β treatment leads to increase of TβRII:TβRI heterodimers rather than heterotetramers [30]. Upon activation, TβRI is relieved of inhibitory effect of the immunophilin protein FKBP12 and actively recruits and phosphorylates the receptor Smads - Smad2 or -3. Smad proteins comprise of three domains, an N-terminal MH1 domain followed by a linked region and a C-terminal MH2 domain. Under resting condition, the MH2 domain and MH1 domain interact with each other. However, receptor-mediated phosphorylation at C-terminal disrupts these interactions exposing N-terminal nuclear localization signal. In addition phosphorylation promotes interaction of MH2 domain with other protein such as Smad4 that as a complex move into nucleus where they regulate large number of genes [31]. In addition to activation of Smad pathway, TGF-β has been shown to elicit non-Smad signaling responses via intermediates such as MAPK, Rho, PI3K-AKT, Src and other signaling molecules (28-31). The remarkable versatility of TGF-β can be attributed to the activation of these non-Smad pathways working in conjunction with Smads to further modulate cellular responses. However, this complexity increases the likelihood of alteration in additional components of this fine-tuned system that ultimately may result in pathological fibrosis.
TGF-β RECEPTOR MEDIATED NON-SMAD SIGNALING
In recent years a significant progress has been made in unraveling the molecular mechanisms involved in the activation of the non-Smad pathways. This topic has been discussed in details in several recent reviews [32-35]. We will discuss selected pathways with a particular relevance to fibrosis.
Erk MAPK Pathway
Activation of Erk MAPK plays an important role in fibrosis by regulating myofibroblast transdifferentiation, cell proliferation and survival, as well as matrix synthesis [36-42] (Fig. 1). Recent studies have provided new insights into the molecular mechanisms involved in activation of this pathway by TGF-β. Lawler et al. [43] were first to demonstrate that in addition to serine/threonine phosphorylation, TβRII undergoes autophosphorylation on tyrosine residues. Further studies from Galliher and Schiemann showed that Src-mediated Tyrosine phosphorylation of TβRII, triggers receptor recruitment of Shc and Grb2 resulting in activation of p38 MAPK signaling [44]. Similar to TβRII, TβRI is also phosphorylated on tyrosine residues in response to TGF-β activation leading to the assembly of Shc/Grb2 complex and activation of Erk1/2 MAPK [45] (Fig. 1). While activation of Erk1/2 MAPK by RTKs (receptor tyrosine kinases) is usually rapid and transient, activation of this pathway by TGF-β is characterized by a prolonged kinetics in dermal fibroblasts [46]. Studies by Samuel et al. have shown that this may be due, in part, to the downregulation of the catalytic subunit of PP2A (protein phosphatase 2A), a key cellular phosphatase that targets Erk1/2 MAPK [46]. SSc fibroblasts were also shown to express reduced levels of PP2A as a result of the constitutive activation of TGF-β signaling. As PP2A is also involved in dephosphorylation of TβRI [47], its downregulation may further contribute to the chronic activation of this pathway in scleroderma fibrosis (Fig. 1). Relevant to this finding, Bandyopadhyay and colleagues [48] have demonstrated that varied expression levels of TGF-β receptors might explain tissue-selective activation of Erk1/2 pathway by TGF-β. The authors observed that in contrast to epithelial cells, in which TGF-β inhibited Erk1/2 activation, Erk1/2 was induced by TGF-β in dermal fibroblasts and microvascular endothelial cells. On the other hand, activation of Smad2/Smad3 was comparable in all cell types. Further, investigation into the levels of receptors revealed that unlike the TβRI levels, which were similarly expressed in both cell types, the levels of TβRII were expressed at much higher levels in fibroblasts. Additional depletion experiments indicated that Erk1/2 MAPK activation is mediated by TβRII in a TβRI independent manner in fibroblasts. Given the importantance of Erk1/2 in regulating profibrotic gene expression, these studies may explain failure of the TβRI kinase specific inhibitors to fully normalize activated phenotype of SSc fibroblasts [49, 50].
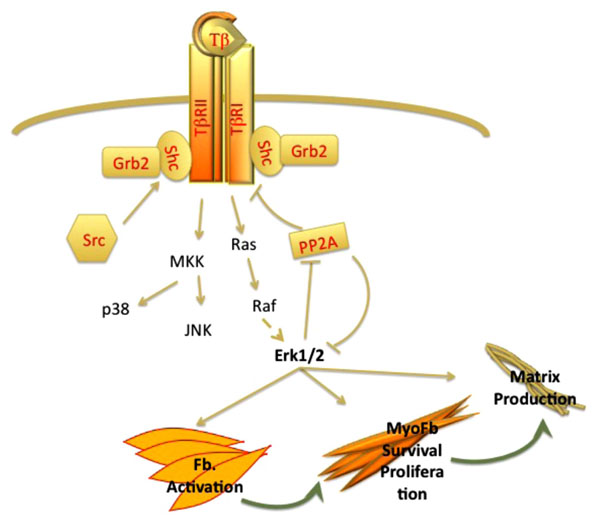
Autocrine regulation of ERK MAPK activation by TGF-β plays a key role in wound healing and fibrosis. TGF-β via Ras-Raf proteins activates Erk pathway. Activated ERK in turn inhibits PP2A, a negative regulator of ERK and TGF-β receptors and stimulates myofibroblast survival and matrix production.
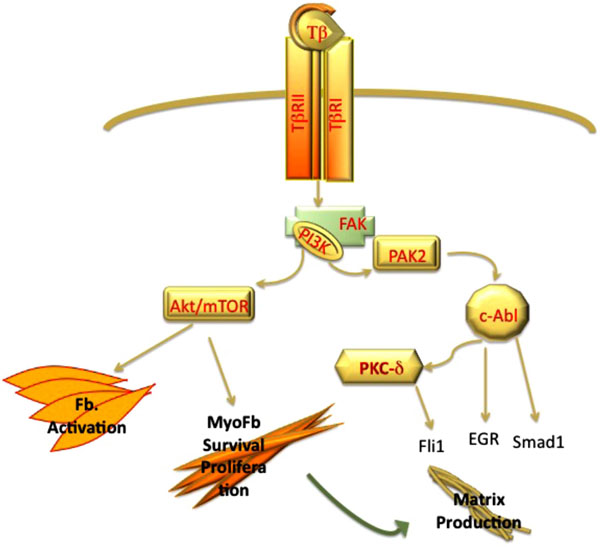
PI3 kinase pathway contributes to TGF-β induced fibrosis via Akt and PAK2 pathways. Activation of Akt/mTOR pathway plays a key role in activation of fibroblast and myofibroblasts survival. PAK2 via c-Abl induces matrix production via activation of EGR and Smad1 pathway and inhibition Fli1 protein.
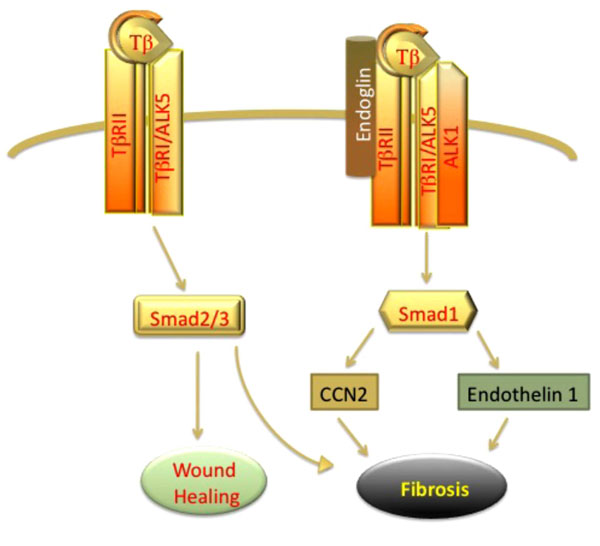
TGF-β activates Smad 2/3 and Smad1 pathway. Activation of classical Smad 2/3 pathway promotes wound healing. Elevated levels of Endoglin in fibroblasts promote Smad1 signaling leading to autocrine upregulation of CCN2 and promoting fibrosis.
PI3K (Phosphoinositide 3-Kinase)-FAK Pathway
Activation of PI3K pathway and its downstream targets plays a central role in the fibrogenic process induced by TGF-β. In contrast to Erk1/2 MAPK, PI3K activation requires both Type II and Type I receptor [51]. The studies by Leof and his colleagues have provided important insights into activation of the TGF-β-PI3K axis in mesenchymal cells. Collectively, these studies demonstrated that TGF-β stimulation leads to recruitment of the p85 subunit of PI3K to FAK (focal adhesion kinase) that acts as a scaffold to organize this signaling complex. Notably, this function of FAK does not require tyrosine kinase activity and is Src-independent [52]. PI3K is a branch point for the activation of the two important profibrotic pathways: PAK2 (p21 activated Kinase)-Abl (Abelson kinase) and Akt-mTOR1 pathways [53] (Fig. 2). The importance of c-Abl pathway has been well established in experimental models of fibrosis, where it was shown that administration of c-Abl inhibitor, imatinib, reduces organ fibrosis [54]. Downstream targets of c-Abl in fibroblasts include such known profibrotic mediators as Egr [55], Smad1 [56, 57], and PKCδ/Fli1 [57]. Furthermore, recent studies have shown that c-Abl-PKCδ pathway may also contribute to the process of endothelial-mesenchymal transition [58]. The Akt-mTOR branch regulates cell proliferation, cell survival, and metabolism [59] (Fig. 2). The importance of the activation of the PI3K pathway in fibrotic disorders is further underscored by the finding that a key negative regulator of this pathway, PTEN, is underexpressed in several fibrotic disorders, including IPF (Idiopathic Pulmonary Fibrosis) [60], scleroderma [61, 62], and liver fibrosis [63].
The important aspect of the TGF-β-PI3K signaling is its role in the process of EMT (epithelial-mesenchymal transition). While EMT has been shown to be a source of collagen producing cells in experimental models of renal and pulmonary fibrosis [64, 65], whether this process contributes to human disease is debatable. This topic has been a subject of recent reviews and will not be discussed herein [35, 59].
Endoglin-Smad1 Pathway in Fibrosis
The type III receptors, betaglycan and endoglin, are part of the TGF-β receptor complex. Structural studies revealed that the cytoplasmic regions of both receptors lack any significant signaling domains and that both receptors are similar in their transmembrane domain [66, 67]. However, these accessory receptors differ in their affinity towards Type I and Type II receptors. Betaglycan facilitates binding of TGF-β isoforms to TβRII, and this effect is most pronounced for TGF-β2, which by itself binds receptor with low affinity [rev in [68]]. Endoglin binds TGF-β1 and TGF-β3, but not TGF-β2, and needs the presence of TGFβRII for binding to TGF-β ligands [rev in [67, 69]]. Endoglin is a critical mediator of angiogenesis and aberrant expression of this co-receptor has been associated with several vascular pathologies including HHT (hereditary hemorrhagic talangiectasias), preeclampsia, and tumor angiogenesis [69, 70]. Studies from the laboratory of Peter ten Dijke have provided important insights into the role of endoglin in the TGF-β signaling in endothelial cells. TGF-β has been shown to utilize distinct receptor subtypes to induce different responses in endothelial cells. TGF-β signaling via ALK5 and subsequent phosphorylation of Smad2/3 is associated with inhibition of cell proliferation and migration, as well as other features consistent with induction of quiescent phenotype, while signaling via ALK1 receptor leads to activation of Smad1/5 and promotion of angiogenic response [67]. It has to be noted that ALK5 is also required for signaling through the ALK1 receptor, although the role of ALK5 in this process is not fully defined. Endoglin is upregulated on proliferating endothelial cells and may function as modulator of a balance between ALK1/Smad1 and ALK5/Smad3 by promoting ALK1 signaling and indirectly inhibiting ALK5/Smad3 signaling. Consistent with this concept, high levels of soluble endoglin are present in patients with preeclampsia and contribute to the defective angiogenesis.
There is increasing evidence that elevated expression of endoglin on mesenchymal cells contributes to the profibrotic TGF-β signaling by triggering endothelial-like signaling via activation of ALK1/Smad1. High levels of endoglin have been demonstrated in liver biopsies and patient serum samples in liver fibrosis [71, 72], renal fibrosis [73, 74], as well as dermal fibroblasts and serum of patients with scleroderma [75-80]. The function of endoglin with respect to regulation of the profibrotic effects of TGF-β has been investigated in several experimental systems. While antifibrotic effects of endoglin were described in some studies [78, 81], majority of the studies support a profibrotic role of endoglin. For example, endoglin was shown to promote profibrotic action of Angiotensin II in cardiac fibroblasts [82]. Furthermore, studies of transdifferentiated HSCs (hepatic stellate cells) have linked endoglin to activation of Smad1/5 signaling and subsequent upregulation of α-SMA [72]. In a subset of scleroderma fibroblasts, constitutive activation of ALK1/Smad1 pathway has been shown to contribute to CCN2 and collagen gene expression [41, 56]. Recent studies by Morris et al. have revealed that this subset of SSc fibroblasts is characterized by the elevated levels of endoglin [61]. Furthermore, endoglin was required for activation of Smad1/5 pathway and collagen and CCN2 gene expression in SSc fibroblasts. Notably, endoglin/ALK1 pathway was also shown to mediate expression of another profibrotic mediator, endothelin-1 in SSc and healthy dermal fibroblasts (Fig. 3). Although, less is known about the potential role of betaglycan in fibrotic disorders, recent studies have suggested that deregulation of this co-receptor may also contribute to fibrosis [83, 84].
CONCLUSIONS
Recent progress in unraveling TGF-β signaling provides a better understanding of the role of this pleiotropic growth factor in fibrosis. While these new information further solidify a central role for TGF-β in development of fibrosis, a critical question regarding suitability of TGF-β as a therapeutic target, remains unanswered. The effective suppression of TGF-β in the affected tissues might be difficult to achieve. Furthermore, serious concerns remain that such a treatment could promote autoimmunity and epithelial hyperplasia [85]. Although anti-TGF-β strategies used to date did not produce toxic effects, this could have been a result of incomplete inhibition of TGF-β, consistent with the absence of beneficial anti-fibrotic effect. The discovery of additional signaling pathways that work in concert with TGF-β in promoting fibrosis may offer new therapeutic options by combining TGF-β suppression with the additional pathway specific inhibition.
ACKNOWLEDGEMENT
The authors were supported by the NIH grants AR42334 and AR44883.
CONFLICT OF INTEREST
Declared none.

