All published articles of this journal are available on ScienceDirect.

The Function of SPARC as a Mediator of Fibrosis
Abstract
Fibrosis is a common end-point of a number of different diseases such as hypertension, diabetes, liver cirrhosis, and those associated with chronic inflammation. Fibrosis is characterized by excessive deposition of extracellular matrix that interferes with normal tissue architecture and function. Increased expression of secreted protein acidic and rich in cysteine (SPARC) in fibrotic tissues has been reported in numerous studies. SPARC is a 43 kDa collagen-binding protein secreted from several different cell types into the extracellular matrix and has been shown to be anti-proliferative and counter-adhesive in vitro. SPARC is a matricellular protein; meaning SPARC is secreted into the extracellular space but does not serve a structural function. Instead, SPARC modulates interactions between cells and the surrounding extracellular matrix. In animal models of fibrotic disease and in human fibrotic tissues, elevated expression of SPARC has been reported in many tissues including heart, lungs, kidneys, liver, dermis, intestine, and eyes. In this review, we will summarize current studies that have examined the expression and functional importance of SPARC in various animal models of fibrosis and in human tissues. Although cellular mechanisms of SPARC in fibrosis remain to be fully elucidated, the studies summarized here provide impetus to further explore the efficacy of SPARC as a potential target for reducing fibrosis.
INTRODUCTION
Secreted protein acidic and rich in cysteine/osteonectin/ BM-40 (SPARC) is a 43 kDa protein secreted from multiple cell types into the extracellular matrix (ECM). SPARC is characterized as a matricellular protein, meaning SPARC is secreted into the extracellular space but does not serve a structural function in the ECM [1]. Instead, SPARC facilitates interactions between cells and the ECM. SPARC is most highly expressed during development as cells are establishing the surrounding ECM architecture and during tissue remodeling, i.e.: wound healing and fibrosis. Cell types expressing SPARC include endothelial cells [2], fibroblasts [3], pericytes [4], astrocytes [5-7], osteoblasts [8, 9], and macrophages [10]. Pertinent to fibrosis, SPARC, a collagen binding protein, has been demonstrated to affect both collagen ECM assembly and the activity of transforming growth factor (TGF)-β.
Expression of SPARC is tightly associated with ECM assembly and is therefore expressed highly in developing tissues and in tissues that undergo high rates of ECM turnover. SPARC was shown to induce cell rounding in endothelial cells, fibroblasts, and smooth muscle cells and thus has been designated as a counter-adhesive protein [11]. SPARC was also shown to be anti-proliferative when added to endothelial cells [12], smooth muscle cells [13], and mesangial cells [14] in vitro. SPARC peptides have been used to characterize the collagen binding capacity of SPARC. Domain four of this four-domain protein, the E-C domain, conferred SPARC binding to collagens I, II, III, and V [15]. The binding site used on fibrillar collagens by SPARC was recently shown to coincide with that of discoidin domain receptor (DDR) 2, a collagen receptor with tyrosine kinase activity [16]. Experiments in cardiac and dermal fibroblasts demonstrated that in the absence of SPARC, collagen accumulated at the cell surface suggesting SPARC was required to promote collagen disassociation from cells [17, 18]. Hence, perhaps increases in cell-associated collagen reflected increased engagement of cell-surface collagen receptors such as DDR2 in the absence of SPARC (Fig. 1).
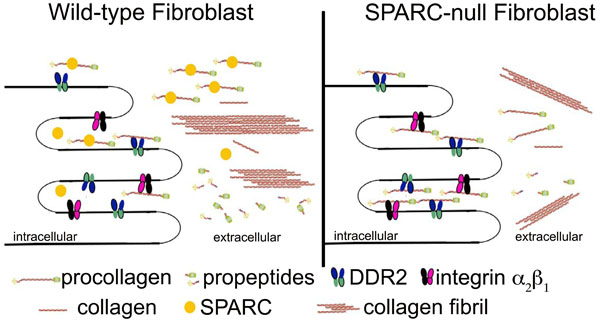
Hypothetical Model of Cellular Mechanisms of SPARC. SPARC binds procollagen as it is secreted from the cell (or procollagen is secreted bound by SPARC) and prevents interaction of procollagen with cellular receptors, such as DDR2 and integrin α2β1 (or others). Therefore, procollagen is appropriately processed and incorporated into collagen fibrils. In the absence of SPARC, procollagen accumulates at the cell surface and is inefficiently incorporated into the collagenous ECM, resulting in less total collagen and fewer thick collagen fibers.
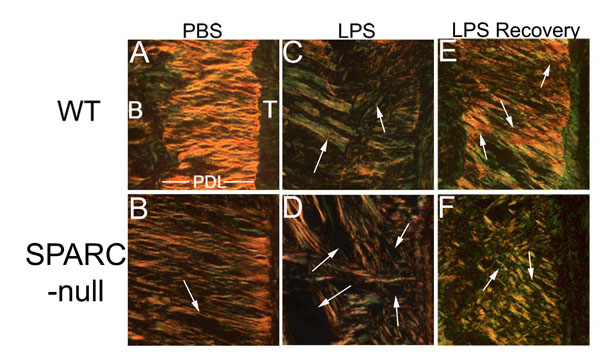
SPARC-null Periodontal Ligament (PDL) is More Susceptible to Collagen Loss Following Lipopolysaccharide Induction of Inflammation. Mice were injected 3 times weekly for four weeks with lipopolysaccharide (LPS, C and D) or vehicle control: phosphate buffered saline (PBS, A and B). SPARC-null PDL (D) exhibited significant loss of collagen as compared to WT (C), white arrows (see ref. [28]). Furthermore, given one week to recover (no LPS injections), WT PDL (E) re-established baseline levels of collagen characterized by the presence of thick collagen fibers (orange/red-white arrows), whereas SPARC-null PDL (F) did not exhibit a return to base-line conditions as indicated by the abundance of thin collagen fibers in SPARC-null mice after one week of recovery (green- white arrows). B: bone, T: Tooth, PDL: Periodontal Ligament.
TGF-β is a cytokine shown to stimulate collagen type I synthesis and SPARC expression [3]. Interestingly, SPARC was also shown to induce TGF-β and collagen I synthesis which suggested a reciprocal regulatory mechanism between SPARC and TGF-β [19]. In support of this, decreases in TGF-β expression, mediated by siRNA silencing, decreased expression of SPARC whereas decreased SPARC expression led to decreased TGF-β activity [20]. As TGF-β is a known regulator of fibrosis, the capacity of SPARC to influence TGF-β signal transduction pathways suggests that SPARC might function, at least in part, to regulate TGF-β activity in fibrotic tissues.
Another useful tool to examine the role of SPARC in ECM synthesis is SPARC-null mice. SPARC-null mice have been established and have been used in a number of different studies. The mouse line most frequently employed has a deletion in the SPARC gene at exon 4, resulting in global abrogation of SPARC expression [21]. Upon gross examination, SPARC-null mice displayed no overt phenotype. However, upon closer examination, a number of abnormalities were detected. For example, these mice developed cataracts at an early age due to aberrant basement membrane integrity formed by the lens epithelial cells [21]. In fact, the majority of distinguishing attributes characteristic of SPARC-null mice has been found to be manifest in ECM structure and function underscoring the important role of SPARC in collagen deposition.
The phenotype of SPARC-null mice suggested that perhaps the primary physiological function of SPARC was tied to collagen binding and to the regulation of ECM assembly and turnover (Fig. 1). Specifically, SPARC-null mice were shown to have significantly less collagen as measured by collagen volume fraction and/or hydroxyproline analysis in skin [22], bone [23], heart [24, 25], and periodontal ligament [26]. Additionally, upon induction of fibrotic stimuli, these mice consistently demonstrated a muted fibrotic response in comparison to WT controls [27]. For example, lipopolysaccharide injected into the gingiva of WT and SPARC-null mice induced an inflammatory periodontal disease in both genotypes [28]. However, SPARC-null mice lost significantly more collagen in the periodontal ligament than the WT controls despite having a decreased inflammatory infiltrate [28] (Fig. 2). Hence, there is a persuasive amount of data that establish the requirement of SPARC expression for a robust fibrotic response.
Fibrosis is a frequent result of diseases such as hypertension, diabetes, liver cirrhosis, and inflammatory processes. During fibrosis, there is deregulated secretion and deposition of ECM that results in tissue remodeling that often interferes with normal tissue function. SPARC expression and up-regulation has been reported in multiple types of fibrosis, both in human tissues and in animal models of fibrotic disease. Additionally, several studies have now shown that inhibition of SPARC expression decreases fibrosis. The purpose of this review is to highlight recent work on the involvement of SPARC in pulmonary, renal, hepatic, dermal, and intestinal fibrosis, as well as in glaucoma. Reviews discussing the role of SPARC in cancer [29] and cardiac fibrosis [27] were recently summarized and are therefore not discussed in detail in this review.
PULMONARY FIBROSIS
SPARC expression is undetectable in adult human lungs [30]. However, in patients with pulmonary fibrosis, the expression of SPARC was found to be increased and was localized to the cytoplasm of pulmonary fibroblasts and within Masson bodies - regions of connective tissue within the air space that are found coincident with pneumonia [30]. Desquamative interstitial pneumonia involves inflammation and tissue turnover, but pulmonary fibroblasts do not typically invade the air spaces. Interestingly, SPARC was not highly expressed in desquamative interstitial pneumonia. These data suggested that SPARC expression was not exclusively due to the turnover of lung tissue, but instead, was involved in the pathological fibrotic process that occurs when pulmonary fibroblasts invade the airspaces [30]. Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic disease [31]. Chang et al. demonstrated that SPARC was overexpressed 8-fold in IPF fibroblasts. In addition, these fibroblasts maintain an overactive, myofibroblast phenotype, as indicated by maintaining α-smooth muscle actin staining through multiple passages in vitro [31]. Utilizing fibroblasts isolated from IPF patients, it was determined that SPARC activated nuclear localization of β-catenin. Furthermore, the activation of β-catenin in IPF fibroblasts promotes stabilization of the myofibroblast phenotype and an anti-apoptotic phenotype characterized by resistance to plasminogen-induced apoptosis and elevated plasminogen activator inhibitor-1[31]. Although the importance of SPARC expression was shown to be relevant in human pulmonary fibrotic disease, the functional significance of increased SPARC expression in fibrotic lungs was more readily addressed using mouse models of pulmonary fibrosis and transgenic SPARC-null mice.
There have been four reports of pulmonary fibrosis induced in WT and SPARC-null mice. Each report used bleomycin to induce pulmonary fibrosis. In this model, bleomycin is intratracheally transfused or transtracheally injected causing a fibrotic response. Strandjord et al. [32] used 0.0035 U/g (~0.075 U/mouse) of bleomycin sulfate administered via a transtracheal injection. In this study, WT and SPARC-null mice had similar distributions of patchy fibrosis. WT lungs injected with bleomycin had increased SPARC expression at both the level of mRNA expression and protein production. Amounts of total collagen were measured by hydroxyproline analysis of the lungs of WT and SPARC-null bleomycin and saline-treated mice. WT bleomycin-treated lungs had significantly greater levels of hydroxyproline than the other groups, suggesting the highest fibrotic response occurred in WT mice. SPARC-null bleomycin injected lungs had slightly elevated hydroxyproline content as compared to the SPARC-null saline-treated group, although this change was not statistically significant. From this study, the authors concluded that SPARC promotes pulmonary fibrosis as indicated by the greater levels of collagen within the lungs of WT mice as compared to SPARC-null mice [32].
Savani et al. [33] and Sangaletti et al. [10] used significantly greater concentrations of bleomycin sulfate infused intratracheally at 0.15 U/mouse. Interestingly, these two studies had divergent outcomes. Both groups reported SPARC-null mice had increased tissue destruction as indicated by greater distortion of lung architecture, including areas with collapsed lung space, and increased inflammatory cell recruitment, specifically neutrophils [33] and T-cells [10], in comparison to bleomycin-treated WT mice [10, 33]. Savani et al. reported SPARC-null bleomycin treated lungs had greater collagen deposition as measured by hydroxproline analysis as compared to WT mice [33]. However, Sangaletti et al. demonstrated WT mice had increased collagen deposition as compared to SPARC-null lungs, also measured by hydroxyproline analysis [10]. The reasons behind these divergent findings are not readily apparent but may include differences in age and/or gender of the mice or variations in methods between the laboratories.
Sangaletti et al. investigated further the role of SPARC in the altered inflammatory response detected between WT and SPARC-null mice [10]. To address this question, SPARC-null bone marrow was transfused into irradiated WT mice (SPARC-null>WT), and vice-versa (WT>SPARC-null) to generate bone marrow chimeric mice. The SPARC-null>WT and WT>SPARC-null chimeric mice were then intratracheally infused with bleomycin to induce pulmonary fibrosis [10]. Interestingly, the SPARC-null>WT had decreased inflammatory infiltrate, as compared to SPARC-null bleomycin lung tissue, and had more severe fibrosis as compared to bleomycin-treated WT lung tissue. WT>SPARC-null maintained a decreased fibrotic response to bleomycin as compared to WT, while having decreased inflammation as compared to SPARC-null. This elaborate study suggested differential functions of SPARC that are cell type dependent and have independent effects on the pathogenic response to disease [10].
Finally, in a study by Wang et al. [34], pulmonary fibrosis was induced in WT mice using 3.5 U/kg (~0.075 U/mouse) of bleomycin infused intratracheally. Following bleomycin infusion, a group of WT mice were intratracheally treated with SPARC siRNA at day 2, 5, and 12 following bleomycin infusion. WT mice exposed to bleomycin developed significant amounts of fibrosis, as measured by Masson’s trichrome stain and by the Sircol colorimetric assay for soluble collagen. However, when the bleomycin exposure was followed by SPARC siRNA, the degree of fibrosis was drastically reduced, although at levels above that of the saline control groups [34]. These data support the findings by Strandjord et al., likely due to the similar concentration of bleomycin used by both groups.
The sum of these studies implicated an important role of SPARC in mediating pulmonary fibrosis, and importantly, those of Wang et al. suggested that inhibition of SPARC expression in a WT background is sufficient to reduce lung fibrosis in response to bleomycin. The seemingly contradictory findings of Savani et al., might reflect additional functions of SPARC in lung tissue with extensive damage. Chang et al. found that in human idiopathic pulmonary fibroblasts, increased SPARC expression was associated with nuclear localization of β-catenin that resulted in decreased apoptosis of myofibroblasts [31]. Sangaletti et al. used bone marrow chimeric mice to demonstrate that the expression of SPARC in pulmonary fibroblasts promoted collagen deposition, while the lack of expression of SPARC in bone marrow cells promoted elevated inflammatory infiltrates [10]. Notably, the combination of SPARC-null>WT had greater levels of fibrosis than WT mice alone, demonstrating the intricate relationship between fibrosis and inflammation [10]. Furthermore, these studies indicate that future attempts to decrease fibrosis by inhibiting SPARC expression should consider the specific cell types to be targeted to prevent undesired consequences of increased inflammation.
RENAL FIBROSIS
In human kidney, SPARC exhibited distinct patterns of expression during development, in mature, and in injured kidneys. In developing kidney, expression of SPARC was localized to the apical side of visceral epithelial cells adjoining the capillary basement membrane of glomeruli [35]. In adult kidney, glomerular SPARC expression was maintained in the visceral epithelial cells but expanded to include parietal epithelial cells, the collecting duct epithelium, smooth muscle cells of arteries, interstitial cells, and the urothelium [35]. Interestingly, serum levels of SPARC were found to be increased in fibrotic renal injury patients [36]. In addition, in renal chronic allograft rejection, SPARC expression by interstitial myofibroblasts was also detected in infiltrating inflammatory cells and in fibrotic regions [35].
A number of studies have been carried out to characterize the expression and potential role of SPARC in renal fibrosis. Pichler et al. [37] characterized SPARC expression in four rat models of renal fibrosis: passive heyman nephritis (PHN), cyclosporine nephropathy (CsA), the remnant kidney model (RK), and angiotensin II infusion. In the normal rat kidney, SPARC is expressed in the glomeruli [38] by the visceral glomerular epithelial cells and is notably absent from the interstitium [37]. In all four models of renal fibrosis, elevated levels of SPARC protein were detected. With PHN there was an increase in SPARC expression in the glomerular epithelial cells, and an increased amount of SPARC in the interstitium localized to spindle shaped cells secreting ECM. In CsA fibrosis, SPARC expression was increased around the dilated and atrophic tubules. RK and angiotensin II models both induced an increase in SPARC expression localized to the interstitial cells. Interestingly, in all models, cells expressing SPARC within the interstitium co-expressed α-smooth muscle actin, a marker for activated fibroblasts and each of these models demonstrated increased interstitial collagen type I. Frequently in fibrosis, a spike in cell proliferation proceeds fibrotic deposition of ECM that is then followed by a decrease in cell proliferation associated with robust ECM secretion and deposition. In each of the four models of renal fibrosis discussed here, cell proliferation began to decrease coincident with maximal increases in SPARC expression [37].
Another model of renal fibrosis and disease is a subtotal nephrectomy model of noninflammatory progressive renal disease, which is characterized by glomerulosclerosis and tubulointerstitial fibrosis [38]. In this rat model, two-thirds of the left kidney is infarcted. In normal rat kidney, SPARC mRNA was localized to glomeruli, however in diseased kidney, mRNA encoding SPARC increased in glomeruli and at the site of tubulointerstitial injury in the infarcted rats. The diseased rats developed hypertension, proteinuria, and renal impairment, all features of renal fibrotic disease. The blockade of the renin-angiotensin system decreased renal fibrosis induced by infarction and also reduced SPARC expression [38].
Expression of SPARC was also increased in rats in response to angiotensin II dependent hypertension, whereas, blockage of angiotensin II resulted in reduced SPARC expression coupled with decreased fibrosis. Socha et al. [39] looked for a direct effect of angiotensin II on expression of SPARC using renal cells isolated from humans and mice. In normal WT mice, glomeruli had faint SPARC localization [40]. Interestingly, a dose dependent increase in both SPARC mRNA and protein levels were found following angiotensin II stimulation in human and murine renal cells. When treated with angiotensin II, both WT and SPARC-null mice had significant increases in blood pressure. However, no significant differences in blood pressure were detected between WT and SPARC-null mice following angiotensin II administration [39]. SPARC-null mice exhibited decreased perivascular and tubulointerstitial fibrosis as compared to WT mice in response to angiotensin II infusion. Decreased fibrosis was associated with decreased TGF-β urinary excretion, significantly less mRNA encoding MMP-2 and MMP-14, and decreased levels of reactive oxygen species (ROS) in SPARC-null versus WT mice. The authors concluded that SPARC expression is stimulated by angiotensin II and that SPARC expression mediates, at least in part, angiotensin II-induced increases in renal fibrosis [39].
Diabetic nephropathy is a state of renal fibrosis that occurs in patients with diabetes and is a major cause of death for those with diabetes. WT and SPARC-null mice were intraperitoneally injected with streptozocin (STZ) to induce diabetes [41]. Six months after the injections, the mice were euthanized and the kidneys were examined. Firstly, differences in survival were noted, 13 of 17 SPARC-null mice survived the entire 24-week study (~76%), while only 7 of 12 WT mice survived the duration of the study (~58%). Both WT and SPARC-null had similar indications of diabetes, as measured by blood urea nitrogen with no significant differences between genotypes. WT diabetic mice had an increase in the mesangial matrix, increases in collagen IV and laminin staining, and increased scoring of glomerulosclerosis. In addition, SPARC expression was increased in the glomerulus and in parietal epithelial cells but not in mesangial cells compared to WT sham mice. In contrast, SPARC-null diabetic mice had no increases in mesangial matrix, decreases in collagen IV and laminin staining within the mesangium, and no detectable changes in the vasculature. Interestingly, both the WT and SPARC-null diabetic mice had increased levels of α-smooth muscle actin expression in the smooth muscle cells of muscular arteries, the interstitial cells, and in sporadic glomeruli [41].
Expression of α-smooth muscle actin is a hallmark of the myofibroblast phenotype, frequently associated with elevated collagen production [42]. Whereas SPARC-null mice had no significant increases in mesangial matrix [41], elevated numbers of myofibroblasts were reported. This finding suggested that SPARC was also required for collagen assembly, not exclusively for differentiation and/or survival of myofibroblasts as indicated by Chang et al. in studies of pulmonary fibrosis [31]. Notably, tubular epithelial cells with no association with increased matrix deposition, were not positive for α-smooth muscle actin expression. Lastly, SPARC-null mice were reported to have decreased levels of TGF-β mRNA [41]. Considering both WT and SPARC-null mice had similar severity of diabetic condition, the differences in survival are likely related to the increased fibrosis in the glomeruli noted in WT versus SPARC-null mice. Also, the decreased expression of TGF-β in this model suggested that in WT mice, SPARC promoted TGF-β synthesis during diabetic nephropathy.
The most recent work characterizing SPARC in renal fibrosis examined the role of SPARC in crescentic glomerulonephritis [40]. Here, 12-week old mice were injected intraperitoneally with an anti-glomerular antibody. This passive nephrotoxic nephritis model was induced in both WT and SPARC-null mice. Nephrotoxic nephritis is characterized by glomerular injury and a reduction in podocyte number. Following disease progression, podocytes had significantly increased SPARC expression. WT and SPARC-null mice had equal podocyte numbers prior to disease onset, but with glomerular injury, WT mice lost significant amounts of podocytes whereas SPARC-null mice did not. In addition, WT glomeruli had effacement of the foot processes of the podocytes and excessive matrix deposition causing damage to the capillary loops. Two markers of podocyte loss, nephrin and podocin, were increased in WT glomeruli and decreased in SPARC-null glomeruli. Mechanical stress or trypsin was used to induce detachment of podocytes in vitro. SPARC-null podocytes were resistant to detachment, an activity that was restored to that of WT by transfection of SPARC cDNA [40].
In each of these rodent studies, expression of SPARC was detectable, although low, in normal kidneys. However, with induction of renal fibrosis by various methods, expression of SPARC increased coincident with fibrosis. In rodents with decreased or abrogated expression of SPARC, the severity of fibrosis was reduced as compared to WT controls. Furthermore, different mechanisms were suggested in each of these studies, emphasizing the potential complexity of SPARC action and highlighting the need for future mechanistic studies of SPARC in renal fibrosis.
HEPATIC FIBROSIS
In normal human tissue sections of liver, weak expression of SPARC has been found associated with spindle cells and in the sinusoidal cells of the periportal areas. In fibrotic liver, SPARC positive spindle cells were contained in fibrotic bands [43]. The up-regulation of SPARC expression in fibrotic human liver was associated with increased levels of collagen I [44]. Nakatani et al. [45] conducted a separate study wherein SPARC expression was evaluated in human liver samples from patients with liver cirrhosis, biliary cirrhosis, and hepatocellular carcinoma. Each of these conditions results in hepatic fibrosis. Similar to previous findings, normal livers exhibited SPARC expression in fibrous structures, identified as nerve fibers, and in the sinusoidal cells. During chronic hepatitis, SPARC expression was more diffusely localized and found in sinusoidal cells in the periportal, intermediate, and pericentral zones of the liver. Hepatic cells expressing SPARC were identified as myofibroblasts, determined by α-smooth muscle actin staining, or as hepatic stellate cells, identified by lipid droplets. Kupffer cells, sinusoidal epithelial cells and hepatocytes were negative for SPARC immuno-localization. SPARC was also found to be a secreted product in conditioned media of human liver myofibroblasts [45].
As SPARC mRNA expression has also been reported in normal and fibrotic livers in mouse and rat models, experiments to address whether reductions in SPARC expression might limit fibrosis have been performed. Camino et al. [46] used thioacetamide (TAA) to induce liver fibrosis in rats in conjunction with adenovirus expressing siRNA against SPARC to decrease SPARC expression. TAA was administered at 600mg/kg/week for 7 weeks simultaneously with SPARC siRNA adenovirus for the first and second TAA treatments. Samples were collected at day 2, day 10, and week 7 to measure SPARC expression and induction of fibrosis. The adenovirus expressing siRNA caused a 50% reduction in SPARC expression in the injected rats. At the completion of 7 weeks with TAA, the level of SPARC within the livers was higher in the adenovirus-free group than in the adenovirus siRNA treated group. The portal-portal fibrous linkages, central-portal fibrous linkages, distal liver architecture and the amount of bile duct proliferation were used to determine the degree of liver fibrosis. The group receiving SPARC siRNA adenovirus had less fibrosis in response to TAA, with fewer thick fibers and a greater proportion of thin and immature fibers, as measured by picrosirius red staining, in comparison to the adeno-GFP and no adenovirus controls [46]. In addition, hydroxyproline analysis confirmed less total collagen in the livers of rats treated with SPARC siRNA adenovirus. The livers of the SPARC siRNA adenovirus group also had significantly less TGF-β secretion as compared to adeno-GFP and no adenovirus controls. In conclusion, the reduction in SPARC expression in the TAA treated rats decreased the degree of fibrosis in these animals, likely due to decreases in levels of TGF-β [46]. Of interest for future experiments is whether inhibition of expression of SPARC by siRNA might be used at later time points to reverse fibrosis in this model. These studies demonstrated that inhibition of SPARC expression decreased hepatic fibrosis however the anti-fibrotic mechanism of SPARC was not addressed.
In a follow-up study, hepatic stellate cell lines from rat and human were used to determine specific cellular mechanisms for the observed decrease in fibrosis in rats injected with SPARC siRNA adenovirus [47]. The authors chose to study hepatic stellate cells because this cell type becomes activated during liver fibrosis. Here, SPARC siRNA was used to decrease SPARC expression and various properties of the hepatic stellate cells were evaluated. Decreased SPARC expression promoted cell adhesion to fibronectin and increased overall cell: cell adhesion, likely dependent upon observed increases in cadherin E expression, thereby decreasing cell migration. Examination of the actin stress fiber network revealed decreased actin and phalloidin staining in SPARC siRNA treated hepatic stellate cells. Reduced SPARC expression also decreased endogenous TGF-β gene expression and protein levels, and prevented responsiveness to exogenous TGF-β. The authors concluded that the decrease in SPARC expression in hepatic stellate cells caused these cells to lose their mesenchymal properties, as indicated by decreased migration, increased adhesion, decreased collagen mRNA, and decreased sensitivity to TGF-β [47].
A separate study has also indicated a connection between SPARC expression and mesenchymal cell phenotype [48]. The melanoma cell line LBLAST and a separate line expressing 80% less SPARC, L2F6, were shown to have differential expression of E-cadherin expression. The LBLAST cell line had decreased E-cadherin and increased N-cadherin, while the L2F6 line had increased E-cadherin and decreased N-cadherin [48]. The transition to N-cadherin expression is indicative of an epithelial to mesenchymal transition. The expression of N-cadherin in these two cell lines was shown to be dependent upon expression of both SPARC and collagen I. In addition, elevated N-cadherin expression was tied to increased invasiveness and transwell migration [48]. These two studies suggested a function of SPARC in EMT and in the maintenance of a mesenchymal phenotype. However, the involvement of EMT in fibrosis is still under debate and further research is needed to fully elucidate the role of SPARC and EMT in fibrosis [49, 50].
DERMAL FIBROSIS
Dermal fibroblasts are frequently used in studies of fibrosis because of 1) their relevance to scleroderma, an autoimmune disease associated with fibrotic deposition of collagen, 2) the need for improved treatments of scar formation, and 3) the relative ease of obtaining primary cells for culture experiments, among other reasons [51, 52]. Circulating levels of SPARC and thrombospondin, another matricellular protein, were found to be elevated in patients with systemic sclerosis (advanced scleroderma) compared to controls [53]. SPARC-null mice have significantly less collagen in the dermis, with thinner collagen fibrils, and increased adiposity [18, 54]. In human fibroblasts, siRNA inhibition of SPARC expression decreased collagen type I synthesis in response to TGF-β stimulation in comparison to normal control fibroblasts [20]. These studies have suggested that a decrease in SPARC expression might lead to decreased fibrosis in diseases such as scleroderma.
Scleroderma affects the dermis, as well as other tissues, and SPARC has been found to be over-expressed in scleroderma fibroblasts [55]. Importantly, by decreasing SPARC expression in vitro using siRNA in human scleroderma fibroblasts, the amount of collagen produced by these cells was decreased by 69% in comparison to non-transfected scleroderma fibroblasts. In addition to the robust reduction in collagen synthesis, the authors also reported slight changes in levels of TGF-β receptor1 and smad3, a downstream signaling molecule in the TGF-β pathway, in SPARC siRNA treated cells, although changes in the levels of these proteins in siRNA-treated cells did not reach statistical significance [56].
Wang et al. [34] examined the role of SPARC in dermal fibrosis by utilizing SPARC siRNA in conjunction with a dermal bleomycin mouse model of fibrosis. Specifically, to induce dermal fibrosis, mice were subcutaneously injected daily for four weeks with 100 μL bleomycin. Three hours following each injection, a group of mice were injected with SPARC siRNA within the same region. Bleomycin induced dermal fibrosis, as measured by Masson’s trichrome and quantification of soluble collagen levels. The mice that received both bleomycin and SPARC siRNA developed significantly reduced levels of dermal fibrosis, although the soluble collagen and immunohistochemical stain of insoluble collagen was slightly higher than untreated control. One would not anticipate complete abrogation of fibrosis mediated by SPARC siRNA as there was likely incomplete knockdown of SPARC [34]. Although the authors confirmed the efficacy of siRNA treatment by measuring levels of SPARC gene expression, immunofluorescent localization of SPARC protein in the areas of fibrosis in the bleomycin treated mice versus regions from the bleomycin + SPARC siRNA mice, would have added to the results. Nonetheless, this important study demonstrated that a reduction in SPARC expression was sufficient to substantially reduce dermal fibrosis in this model and further support the use of strategies that inhibit SPARC production in the treatment of fibrosis.
INTESTINAL FIBROSIS
Crohn’s disease is a human condition characterized by an intestinal obstruction due to excessive ECM deposition. A mouse model of Crohn’s Disease uses trinitrobenzene sulfonic acid (TNBS) that results in increased expression of matrix metalloproteinase (MMP) 1 and collagen type I, in the absence of increased MMP 3 and collagen type III - a protein profile consistent with the relative expression of ECM proteins in Crohn’s Disease. Klopcic et al. [57] examined the role of a pro-fibrotic and anti-fibrotic compound, indomethacin and retinoic acid (RA) respectively, in relation to the expression of SPARC in the TNBS fibrosis model. Mice were split into three groups: TNBS only, TNBS and indomethacin, or TNBS and RA. The authors showed that the TNBS-indomethacin group had the most severe fibrosis, followed by the TNBS-only and TNBS-RA groups. Interestingly, SPARC expression at 2-weeks was greater in TNBS- only mice with lower fibrosis than in TNBS-only mice with higher levels of fibrosis. However, when TNBS and indomethacin were jointly administered, SPARC expression was highest versus the other groups at 2-weeks. The TNBS-RA group also had higher SPARC expression at 2-weeks than the TNBS-high fibrosis group. The authors concluded from these results that SPARC might serve an anti-fibrotic function in the intestine, as the TNBS-only high fibrosis group had the lowest level of SPARC. These results demonstrated there are potential tissue-specific mechanisms of SPARC activity in fibrosis [57]. However, because this study found only an association of histological levels of SPARC protein and levels of SPARC mRNA in the absence of a mechanistic study, it is difficult to extrapolate whether SPARC truly had an anti-fibrotic activity in the intestine. Instead, we can conclude that different agents likely lead to fibrosis through distinct pathways and further experiments to identify causality of SPARC expression in intestinal fibrosis are needed. Future studies could compare the effects TNBS and indomethacin in SPARC-null versus WT mice, and could also utilize SPARC shRNA to knockdown SPARC expression following TNBS and indomethacin treatment to determine direct effects of SPARC on intestinal fibrosis.
GLAUCOMA
In human eyes, Tenon’s capsule becomes irreversibly scarred by glaucoma. Scarring is similar to other types of fibrosis in that there is abundant ECM secretion and assembly. SPARC expression was found to be up-regulated in scarred tissue in comparison to normal Tenon’s capsules [58]. In both normal and scarred capsules, SPARC expression was localized primarily to blood vessels. Expression of SPARC has been found to be stimulated by TGF-β1 and SPARC has been found to have additive effects with TGF-β1 on cell proliferation. To determine whether the expression of SPARC within the scars of Tenon’s capsule was contributing to fibrosis or caused by fibrosis, fibroblasts from Tenon’s capsule were examined in vitro. TGF-β induced SPARC expression in these fibroblasts that had low levels of basal SPARC expression. In addition, SPARC stimulated proliferation of fibroblasts from Tenon’s capsule, an affect that was additive to that of TGF-β. SPARC was found to enhance collagen gel contraction by Tenon’s fibroblasts, an activity considered critical for scar development. Lastly, TGF-β was also induced by SPARC in these cells [58]. Hence, in fibroblasts from Tenon’s capsule, SPARC and TGF-β appear to have reciprocal and synergistic activities that promote cell proliferation and tissue contraction.
Currently, following glaucoma surgery, mitomycin-C is used to prevent scar formation, but this treatment is not completely effective and, often times, scar formation prevails. To test whether SPARC was a critical factor in Tenon fibroblast function, siRNA against SPARC was performed [59]. Inhibition of SPARC by siRNA in human Tenon’s fibroblasts decreased necrotic cell death, decreased fibroblast migration in a scratch assay, decreased collagen gel contraction, and prevented collagen I and fibronectin induction by TGF-β2. Finally, expression of several MMPs was also significantly decreased in the presence of SPARC siRNA treatment [59]. These results suggested that SPARC suppression might provide an improved treatment option for prevention of scars associated with glaucoma and is an area worthy of further study.
Furthermore, an animal model of wound repair in Tenon’s capsule was used to assess whether SPARC was required for efficient wound healing in mice. In this model, a fistula is created in the anterior chamber of the eye after dissection of the cornea and incision of the sclera [60]. The fistula is then sutured and is visible as a filtering bleb in the conjuctiva. WT mice had a 0% survival rate from the surgery at day 14, while SPARC-null mice had an 87.5% survival rate. The increase in survival in the SPARC-null mice was coincident with decreases in collagen accumulation within the cornea, as detected by picrosirius red staining [60]. Hence, SPARC was shown to mediate both fibrotic collagen deposition and survival in this model of glaucoma.
Mechanisms of SPARC in Fibrosis
Elucidation of the cellular mechanisms of SPARC action on collagen deposition is an active area of research. Possibly, different activities of SPARC might impact distinct phases of fibrotic collagen deposition. As an example, the absence of SPARC has been shown to affect both levels of collagen and the morphology of assembled collagen fibers. The capacity of SPARC to influence TGF-β activity is predicted to affect fibroblast activity including proliferation, migration, and collagen production. Clearly the number of fibroblasts in a fibrotic tissue, increased by either resident cell proliferation or recruitment of fibrocytes, impacts the fibrotic response. Hence, inflammation that often precedes fibrotic events might serve to increase the number and recruitment of fibroblasts. Results reported by Sangaletti et al., suggested that expression of SPARC by inflammatory cells served to dampen the inflammatory response [10]. Hence, the shift from a fibroblast proliferative phase to an ECM deposition phase might be regulated, at least in part, by expression of SPARC. In the event that SPARC enhances TGF-β activity, SPARC would then serve to decrease inflammatory responses through inhibition of TNF-α while promoting ECM deposition by fibroblasts and fibrocytes. However, caution should be made regarding over-simplification of the positive regulation of TGF-β activity by SPARC as notable differences in SPARC activity on TGF-β signaling have been found (reviewed in [61]). Hence, regulation of TGF-β activity by SPARC seems to be, at the very least, cell-type specific. For example, Rivera and Brekken recently reported in pericytes that the absence of SPARC resulted in increased TGF-β activity mediated through interaction of SPARC with the TGF-β receptor endoglin that suggested SPARC negatively influenced TGF-β signaling in pericytes [4]. Given the complexity of TGF-β activation and signaling pathways, possibly SPARC serves to enhance particular TGF-β pathways in certain cell types.
Second to increases in fibroblast number is the assembly of secreted collagen into collagen fibrils in fibrotic tissues. Clearly, the absence of SPARC has been shown to result in collagen fibers with altered morphology. Collagen fibers and fibrils are thinner in SPARC-null mice [22]. Hence, in addition to reduced amounts of collagen, the collagen architecture in SPARC-null tissues has been found to be aberrant both in normal tissues and in response to fibrotic stimuli. Thinner collagen fibers are predicted to be more readily digested by collagenases in the extracellular space. During tissue remodeling, increased activity of ECM degrading enzymes would then lead to increased ECM turnover and perhaps greater instability of ECM structures assembled in the absence of SPARC. The capacity of SPARC to bind to fibrillar collagens is likely a critical factor in the modulation of collagen fibril assembly.
Of interest in this regard is the recent characterization of the SPARC binding site on fibrillar collagen and the knowledge that the site engaged by SPARC is the exact site engaged by the collagen receptor DDR2 [62]. One speculation is that SPARC functions to limit collagen binding to DDR2 and perhaps other collagen receptors, such as integrins (Fig. 1). As collagen engagement by DDR2 has been shown to promote fibroblast proliferation, SPARC would then act as an anti-proliferative agent in this capacity. Notably, SPARC has also been shown to bind β1 integrin in adipocytes and affect integrin-linked kinase (ILK) activity [63]. Thus the capacity of SPARC to influence cell responses to cytokines and ECM, possibly through differential engagement of collagen cell surface receptors, is an intriguing idea and the important subject of future research.
CONCLUSIONS
In conclusion, multiple models of fibrosis have been characterized in terms of SPARC expression and function. Overall, these studies have demonstrated that SPARC expression is closely tied to increased collagen deposition. In all studies to date in which engineered decreases or abrogation of SPARC expression were performed, significant decreases in fibrosis were found with one exception. In one study of pulmonary fibrosis that utilized a particularly high dose of bleomycin, SPARC-null mice had greater fibrosis than WT counterparts. In addition, in one study of intestinal fibrosis, SPARC expression was not strictly correlated with the extent of fibrosis. These two studies demonstrated that, while SPARC is ubiquitously expressed in multiple types of fibrotic diseases, perhaps tissue-specific functions and time-dependent expression of SPARC might result in distinct cellular responses. Nonetheless, there is consistent evidence that decreased expression of SPARC affected fibrotic outcomes and most frequently resulted in decreases in collagen deposition. Therefore, inhibition of SPARC activity is a potential treatment strategy in fibrotic diseases. Additional research is needed in higher order primates to determine if SPARC inhibition would have similar efficacy on fibrosis in humans and to determine whether negative side effects in organs or tissues not affected by fibrosis might be presented. Because of the capacity of SPARC to stimulate TGF-β synthesis and activity in some cell types, it is also plausible that SPARC over-expression could be useful to promote regeneration of tissues that are currently un-reparable, e.g. the periodontal ligament following periodontal disease. However, caution needs to be applied, as increased levels and activity of TGF-β have been shown to have detrimental effects, as well, particularly in developing tissues (e.g. [64]).
At this point, it is well established that expression of SPARC is tightly connected to various fibrotic diseases. Unanswered questions remain as to specific cellular mechanisms of SPARC, and to determine which activities of SPARC might be tissue or cell type specific. A better understanding of cellular activities influenced by SPARC that are physiologically relevant to fibrosis are an active and interesting area of research in this important field.
ACKNOWLEDGEMENTS
This work was supported in part by National Institute of Health [T32DE017551, R25 HL092611, 2P20RR017696, HL094517, R01DE018290] and a Veteran’s Administration Merit Award.
CONFLICT OF INTEREST
Declared none.

