All published articles of this journal are available on ScienceDirect.

Role of MicroRNAs in Fibrosis
Abstract
Fibrosis is the leading cause of organ dysfunction in diseases such as systemic sclerosis, liver cirrhosis, cardiac fibrosis, progressive kidney disease, and idiopathic pulmonary fibrosis. The hallmark of fibrosis is tissue remodeling with excess deposition of extracellular matrix components, predominantly collagens. Different cell types, cytokines, growth factors, and enzymes interact in complex pathogenic networks with myofibroblasts playing a pivotal role. MicroRNAs are small non-coding RNAs acting as negative regulators of gene expression at the post-transcriptional level. MicroRNAs have been associated with many basic cellular processes as well as with a wide spectrum of diseases, most notably cancer. This review provides a comprehensive overview of microRNAs regulating profibrotic pathways and extracellular matrix synthesis. The potential of miRNA for targeted therapeutic approaches in fibrotic disorders is also discussed.
INTRODUCTION
Fibrosis is a leading cause of organ dysfunction in diseases, either as the outcome of an uncontrolled reaction to chronic tissue injury, or as the primary disease itself in predisposed individuals [1-4] (Table 1). Independently from the initiating trigger and the tissue-specific response, the hallmark of fibrosis is excess deposition of extracellular matrix (ECM) components, predominantly collagens and associated glycoproteins. Accumulating ECM replaces functional tissue, and disrupts organ architecture [1]. Different cell types, cytokines, growth factors, and enzymes are orchestrated together in complex pathogenic networks, converging on the activation and/or recruitment of fibroblasts and their differentiation into myofibroblasts, which are α-smooth muscle actin (α-SMA) enriched cells overproducing ECM [5-7]. It is currently believed that along with these cellular key players, a number of regulatory factors also have substantial effects on fibrosis and may be responsible for inter-organ variability of fibrotic manifestations [8].
Examples of Primary (Idiopathic) and Secondary Fibrotic Disorders with Multiple/Single Organ Presentation
| Primary | Secondary |
|---|---|
| Systemic | |
| Systemic sclerosis | Graft versus host disease |
| Drug-induced nephrogenic systemic fibrosis | |
| Sarcoidosis | Secondary amyloidosis |
| Amyloidosis | Post-radiation |
| Toxic environmental exposure | |
| Storage disorders (hemochromatosis, glycogenosis, Gaucher’s disease, etc) | |
| Lungs | |
| Idiopathic pulmonary fibrosis | Pneumoconiosis |
| Histiocytosis X | Infectious pneumonitis |
| Cryptogenic organizing pneumonia | Tuberculosis |
| Hypersensitive pneumonitis | |
| Inherited disorders | |
| Autoimmune diseases | |
| Transplant rejection | |
| Drug-induced | |
| Post-radiation | |
| Sarcoidosis | |
| Amyloidosis | |
| Heart | |
| Idiopathic restrictive cardiomyopathy | Coronary artery disease/myocardial infarction |
| Pressure-overload heart (long standing arterial hypertension, valvular disease) | |
| Infectious myocarditis | |
| Autoimmune diseases | |
| Transplant rejection | |
| Familial hypertrophic cardiomyopathy | |
| Arrhythmogenic right ventricular cardiomyopathy | |
| Drug-induced | |
| Post-radiation | |
| Sarcoidosis | |
| Amyloidosis | |
| Kidney | |
| Idiopathic nephrotic syndrome | Diabetic glomerulosclerosis |
| Idiopathic membranoproliferative glomerulonephritis | Hypertensive nephrosclerosis |
| Autoimmune glomerular diseases | |
| Drug-induced | |
| Post-radiation | |
| Amyloidosis | |
| Transplant rejection | |
| Liver | |
| Primary biliary cirrhosis | Chronic viral hepatitis |
| Primary sclerosing cholangitis | Schistosomiasis |
| Alcoholic liver disease | |
| Nonalcoholic fatty liver disease | |
| Drug-induced | |
| Toxic environmental exposure | |
| Inherited metabolic disorders | |
| Autoimmune hepatitis | |
| Intestinal bypass | |
Major Molecular Mediators Elevated in Fibrotic Disorders
| Cytokines |
| Transforming growth factor-β (TGF-β) |
| Interleukin (IL)-4 |
| IL-13 |
| IL-17 |
| Chemokines |
| CCL2 |
| CXCL8 |
| CXCL12 |
| Growth Factors |
| Connective tissue growth factor (CTGF) |
| Platelet-derived growth factor (PDGF) |
| Vascular endothelial growth factor (VEGF) |
| Fibroblast growth factor (FGF) |
| Insulin-like growth factor-1 (IGF-1) |
| Small Secreted Signaling Proteins |
| Wnt proteins |
| Endothelin-1 (ET-1) |
MicroRNAs Linked to Fibrosis
| MicroRNA | Effect | Organ | Predicted Target and Confirmation Level | Putative Role in Fibrosis | References |
|---|---|---|---|---|---|
| let-7d | Anti-fibrotic | Lung | Reporter gene assay (HMGA2) | ↓EMT | [48] |
| miR-107 | Anti-fibrotic | Kidney | - | ↓ Interstitial fibrosis in allograft transplant | [49] |
| miR-132 | Anti-fibrotic | Heart, liver | Target mRNA changes in mimic/silencing miRNA transfection (RAS-GTPase, MeCP2) | ↑angiogenesis and ↓myofibroblasts differentiation | [50, 51] |
| miR-133 | Anti-fibrotic | Heart | Reporter gene assay (CTGF, TGF-β1) | ↓ TGF-β signaling and ↓CTGF signaling | [52, 53] |
| miR-141 | Anti-fibrotic | Kidney | Reporter gene assay (TGF-β2) | ↓TGF-β and ↓TGF-β dependent EMT | [54] |
| miR-142-3p | Pro-fibrotic | Kidney | - | ↓Interstitial fibrosis in allograft transplant | [49] |
| miR-15b, miR-16 | Anti-fibrotic | Liver, lung | Reporter gene assay (Bcl-2) | ↑apoptosis | [55, 56] |
| miR-150 | Anti-fibrotic | Liver | Target protein changes in mimic/silencing miRNA transfection (c-myb) | ↓activation and proliferation in HSC | [57] |
| miR-155 | Pro-fibrotic | Lung, liver | Reporter gene assay (KGF-FGF-7); target protein changes in mimic/silencing miRNA transfection | ↓mesenchymal-epithelial cross-talk | [58, 59] |
| miR-17-92 cluster (miR-18a, 19a/b) | Anti-fibrotic | Liver, heart, lung | Target protein changes in mimic/silencing miRNA transfection (CTGF; CTGF and TSP-1) | ↓CTGF and ↓ TSP-1 | [48, 60, 61] |
| miR-192 | Pro-fibrotic | Kidney | Reporter gene assay (ZEB1/2) | ↑collagen synthesis and ↑EMT | [62, 63] |
| miR-194 (clustered with miR-215) | Anti-fibrotic | Liver | Target protein changes in mimic/silencing miRNA transfection (rac1) | ↓activation and ↓ proliferation in HSC | [57] |
| miR-199a/b | Pro-fibrotic | Heart, Liver | Reporter gene assay (Dyrk1); | ↑ECM synthesis via calcineurin/NFAT signaling | [64, 65] |
| miR-200 family (miR-200a/b) | Anti-fibrotic | Kidney, liver | Reporter gene assay (TGFβ2 for miR-200a, ZEB1/2 for miR-200b); target mRNA changes in mimic/silencing miRNA transfection | ↓TGF-β signaling and ↓ TGF-β -dependent EMT | [54, 59, 66] |
| miR-204 | Anti-fibrotic | Kidney | - | ↓Interstitial fibrosis in allograft transplant | [49] |
| miR-208 | Pro-fibrotic | Heart | Reporter gene assay (THRAP1 and myostatin) | ↓myocardial contractility and ↑fibrosis by de-repression of negative regulators of stress-response genes | [67] |
| miR-21 | Pro-fibrotic | Lung, heart, kidney | Reporter gene assay (Smad7 in lung); in vivo functional assay (Spry1 in heart); in vitro functional assay for Smad interactions | ↑TGF-β canonical (via Smad7 de-repression of Smad2/3) and non canonical signaling (via Spry1 de-repression of ERK/MAPK) | [68-70] |
| miR-211 | Anti-fibrotic | Kidney | - | ↓Interstitial fibrosis in allograft transplant | [49] |
| miR-215 | Pro-fibrotic | Kidney | Reporter gene assay (ZEB1/2) | ↑EMT | [63] |
| miR-216, miR-217 | Pro-fibrotic | Kidney | Reporter gene assay (PTEN) | ↑Akt signaling via PTEN de-repression | [71] |
| miR-23a (clustered with miR-27a) | Pro-fibrotic | Lung | Systems biology approach and real time PCR (ZEB1) | ↑EMT | [72] |
| miR-26a/b | Anti-fibrotic | Lung | Microarray | - | [48] |
| miR-27b, miR-27a | Pro-fibrotic | Liver | Reporter gene assay (RXR-α) | ↑lipid accumulation and ↑proliferation of HSC | [73] |
| miR-29a/b/c | Anti-fibrotic | SSc, heart, lung, kidney, liver | Reporter gene assays (different collagens, FBN1, ELN1, MMP2, ITGB1; target mRNA changes in mimic/silencing miRNA transfection | Directly ↓ECM synthesis | [47, 48, 59, 74-77] |
| miR-30c | Anti-fibrotic | Heart, lung | Reporter gene assay (CTGF) | ↓CTGF signaling | [48, 51] |
| miR-32 | Pro-fibrotic | Kidney | - | ↓Interstitial fibrosis in allograft transplant | [49] |
| miR-335 | Anti-fibrotic | Liver | Target mRNA changes in mimic/silencing miRNA transfection (TNC) | ↓HSC activation and migration | [78] |
| miR-338* | Pro-fibrotic | Lung | Reporter gene assay (LPA1) | ↑fibroblast proliferation | [79] |
| miR-34a | Pro-fibrotic | Liver | Reporter gene assay (ACSL1) | ↑lipids biosynthesis | [59, 80] |
| miR-377 (clustered with miR-382) | Pro-fibrotic | Kidney | Reporter gene assay (PAK1, SOD) | ↑fibronectin synthesis | [81] |
| miR-382 | Pro-fibrotic | Kidney | Reporter gene assay (SOD2) | ↑EMT mediated by ↓ protection against mitochondrial oxidative stress | [82] |
| miR-449a/b | Anti-fibrotic | Kidney | Target mRNA changes in mimic/silencing miRNA transfection (SERPINE1) | ↑ECM synthesis in hypoxic conditions via downregulation of miR-449 | [83] |
| miR-590 | Anti-fibrotic | Heart | Reporter gene assay (TGFβR2) | ↓TGF-β signaling | [53] |
EMT = epithelial-to-mesenchymal transition; HMGA2 = high mobility group AT-hook 2; RAS-GTPase = Rat sarcoma guanosine triphosphatase; MeCP2 = methylCpG binding protein 2; CTGF = connective tissue growth factor; TGF-β = transforming growth factor- β; Bcl-2 = B-cell lymphoma 2; HSC = hepatic stellate cell; c-myb = avian myeloblastosis proto-oncoprotein; KGF = keratinocytes growth factor; ZEB = zinc finger E-box-binding homebox; rac1 = Ras-related C3 botulinum toxin substrate 1; Dyrk1 = dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1a; NFAT = nuclear factor of activated T-cells; βMHC = β myosin heavy chain; Smad = small mother against decapentaplegic; Spry1 = protein sprouty homolog 1; ERK = extracellular signal-regulated kinase; MAPK = mitogen activated protein kinase; PTEN = phosphatase and tensin homolog; Akt = non-specific serine/threonine-protein kinase; RXR-α = retinoid X receptor-α; FBN1 = fibrillin1; ELN1 = elastin1; MMP2 = metalloproteinase 2; ITGB1 = integrin β1; TNC = tenascin; LPA1 = lipophosphatidic acid; ACSL1 = Long-chain-fatty-acid-CoA ligase 1; PAK1 = p21 activated kinase 1; SOD = superoxide dismutase; TGF-β receptor 2 = transforming growth factor- β receptor type 2; TSP-1 = thrombospondin-1.
In this scenario, microRNAs (miRNAs) may play the role of conductors in the pathogenesis of fibrosis [9]. miRNAs are small, non-coding RNAs with a strictly regulated biogenesis. This is combined with an extremely flexible and sophisticated regulatory function, allowing simultaneous targeting of multiple proteins involved in different crucial biological pathways of specific cell types and tissues [10]. Consequently, miRNAs have been associated with many basic cellular processes such as cell death, proliferation and differentiation. On the other hand, miRNA dysregulation has also been linked to a wide spectrum of diseases, most notably cancer [11].
A number of recent reviews have addressed the role of miRNA in fibrosis with a focus on organ-specific miRNA alteration [12-15]. Here we paid special attention to miRNA alterations common to different fibrotic disorders such as systemic sclerosis (SSc) [16], liver cirrhosis (LC) [17], cardiac fibrosis (CF) [18, 19], chronic kidney disease (CKD) [20], and idiopathic pulmonary fibrosis (IPF) [21]. miRNAs directly regulating ECM synthesis and miRNAs regulating TGF-β/CTGF signaling as a key profibrotic pathway were considered for this review.
MicroRNA BIOGENESIS AND FUNCTION
miRNAs are a class of small, evolutionarily conserved, non-coding RNAs, which repress the expression of protein coding genes at the post-transcriptional level via a partially complementary binding to the 3’ untranslated region (UTR) of target mRNAs ([22], for comprehensive review). To date, 1426 miRNA encoding sequences have been identified in the human genome and more than half of them have been experimentally validated (ftp://mirbase.org/pub/mirbase/CU RRENT/genomes/hsa.gff) [23]. Most miRNA genes are located in introns, exons and UTRs of protein coding genes. Approximately one quarter of miRNAs are encoded in intergenic regions, and display an independent transcription unit, whereas intronic and exonic miRNAs are usually co-transcribed and co-expressed with the host gene [22]. In addition, a half of known miRNAs are encoded by polycistronic units, generating multiple miRNAs, often related in their structure and function [22, 24].
miRNAs are transcribed as long polyadenylated RNAs, pri-miRNAs, which contain one or more mature miRNA sequences. Pri-miRNAs are cut by the endonuclease Drosha and its co-factor DiGeorge syndrome critical region 8 (DGCR8), originating intermediate 60-100 nucleotide hairpin structures called pre-miRNAs [22]. Pre-miRNAs are transported to the cytoplasm by exportin 5 and further processed by another endonuclease, Dicer, releasing functionally active, mature miRNAs with a length of ≈ 22 nucleotides. Each mature miRNA is then incorporated into the RNA-induced silencing complex (RISC), which allows the recognition of target mRNA and the formation of a miRNA:mRNA duplex [22]. The base pairing of the duplex is mediated by a distinct seed region at nucleotides 2-8 of the miRNA, and alters mRNA stability or affects protein translation [25]. Translational repression and also target mRNA destabilization predominate in humans, although in rare cases miRNAs can switch their negative regulatory function towards gene activation by promoting mRNA translation [26].
The “imperfect” miRNA:mRNA binding is the basis for an extremely flexible regulatory system, as one miRNA can regulate hundreds of genes, and one gene containing multiple partially complementary binding sites in its 3’UTR can be regulated by multiple miRNAs [24]. In addition, this flexibility in miRNA:mRNA interaction is further expanded by a number of variables: miRNA binding site accessibility, RNA secondary structure, position of the binding site in the 3’UTR and its distance from the stop codon, and reciprocal interference between different miRNAs and RNA binding proteins sharing the same target [27]. As a consequence, miRNA activity depicts a gray scale rather than a black-or-white regulatory scenario [27, 28]. This aspect is particularly relevant for the regulation of multistep biological processes such as signaling pathways: while the effect of a given miRNA on the overall activity of the pathway can be potent, this often results from the sum of subtle modulations on multiple constituents of the same pathway rather than from strong effects on a single molecule [28]. Comprehensive in silico analysis of miRNA targets is provided at the miRGen database (http://www.diana.pcbi.upenn.edu/miRGen.html), which merges the results of multiple miRNA prediction databases, such as DIANA microT, miRanda (microrna.org), miRanda (miRBase), PicTar (4-way and 5-way), and TargetScanS [29].
Despite this versatile regulatory activity, the expression of miRNA genes is tightly controlled, and specific cell types often display a certain miRNA profile [22]. Regulation of miRNA expression at both the transcriptional and post-transcriptional level is still poorly understood. However, multiple mechanisms may play a role. At the transcriptional level, miRNAs can control their own expression establishing either positive or negative regulatory loops via an indirect regulation of transcriptional activators or repressors [30]. Epigenetic modifications such as DNA and histone methylation and histone deacetylation also participate in the modulation of miRNA transcription [31, 32]. At the post-transcriptional level, factors influencing the activity of the endonucleases Drosha and Dicer affect selective miRNA cluster maturation [33, 34]. In addition, some pseudogene mRNAs can act as miRNA decoy through a 3’UTR which is highly homologous to the one of the related protein-coding mRNA, thus modulating cell levels of “free” miRNA [35]. Finally, exosome-mediated cell-cell exchange of RNA has also been described as a determinant of cellular miRNA profile [36].
Fig. (1) gives an overview of miRNA biogenesis and function.
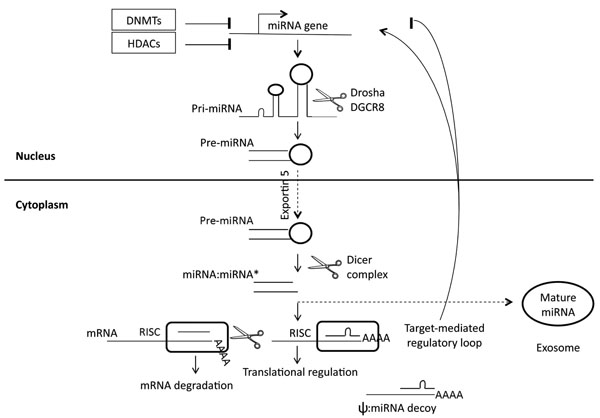
Schematic miRNA biogenesis and regulation. The miRNA gene is transcribed (pri-miRNA) and cut in the nucleus to generate pre-miRNA. Pre-miRNA is transported to the cytoplasm and is further processed there. Mature miRNA is originated as a mature miRNA:miRNA* (passenger strand) duplex. Mature miRNA is incorporated in RISC to bind a target mRNA. Perfect miRNA-mRNA binding leads to mRNA cleavage, while imperfect miRNA-mRNA binding leads to translational repression (rarely to translational stimulation). Levels of cellular bioactive miRNA can be controlled by both transcriptional regulation, including epigenetic modifications and target-dependent loops, and post-transcriptional regulation, including decoy mechanisms exerted by pseudogenes, and intercellular exosomedependent trafficking. DNMTs = DNA methyltransferases; HDACs = histone deacetylases; DGCR8 = DiGeorge syndrome critical region 8; RISC = RNA-induced silencing complex; ψ = pseudogene.
SSC: A MODEL FOR FIBROTIC DISEASES?
Despite fibrotic disorders include a large number of primary and secondary entities with distinct features, major extracellular mediators of fibrosis and related signaling pathways are nevertheless common to most of them. This includes cytokines, chemokines, growth factors, and other small secreted signaling proteins (Table 2) [2].
SSc is a systemic fibrotic disease of unknown origin characterized by the presence of three main pathophysiological pathways in each target organ/system: vascular injury, immune system activation, and interstitial fibrosis [37]. Thus, we propose SSc as a paradigm of fibrotic disorders, based on the following observations: i) fibrosis in SSc can virtually affect all organs/systems, although the disease displays apparent clinical involvement at preferential sites (skin, lungs, heart, kidneys, gastrointestinal tract, muscles); ii) initial factors triggering pro-fibrotic pathways in other fibrotic diseases are also discussed in SSc and SSc-like diseases including infections, autoimmune phenomena, and exposure to toxics or radiations [38-41]; iii) consistent evidence for the activation of major pro-fibrotic pathways common to most fibrotic diseases has been reported in SSc, including transforming growth factor-β (TGF-β) pathways, the platelet-derived growth factor (PDGF) pathway, and, most recently, the canonical Wnt-β-catenin pathway and the serotonin pathway [42-47].
OVERVIEW OF PRO- AND ANTI-FIBROTIC MicroRNAs
According to the current literature, ≈ 40 miRNAs have been linked to fibrosis in various organs and disease settings (Table 3). In general, the miRNA profile is specific for a given cell-type, and even more specific in relation to the developmental and functional organ status [22]. This might explain, why many miRNAs with pro- or anti-fibrotic properties were found to be dysregulated in specific organs and specific fibrotic diseases [48, 49, 52-54, 57, 62, 63, 67, 71-73, 78-83]. On the other hand, some miRNAs have been found to be abnormally expressed in more than one fibrotic disorder [47, 50, 51, 55, 56, 58-61, 64-66, 68-70, 74-77]. Predicted targets of approximately half of miRNAs involved in fibrosis have been validated by reporter gene assay [84]. Most of these miRNAs directly induce or protect from fibrosis by targeting TGF-β canonical and non-canonical pathways [53, 54, 68-71], connective tissue growth factor (CTGF) [51, 52, 60, 61], ECM structural proteins or enzymes involved in ECM remodeling [47, 74-77, 81, 83]. Another set of miRNAs indirectly regulates fibrogenesis by affecting epithelial-to-mesenchymal transition (EMT) [48, 50, 51, 58, 62, 63, 66, 82], or by inducing proliferation and resistance to apoptosis in myofibroblasts [51, 55-57, 73, 78, 80] (Table 3).
MicroRNA REGULATION OF TGF-B/CTGF SIGNALING
TGF-β is a central mediator of fibrosis [1] and it is ubiquitously expressed in tissues [85]. TGF-β also induces expression of CTGF, and CTGF can in turn enhance TGF-β signaling, along with a number of other pro-fibrotic factors including vascular endothelial growth factor (VEGF), endothelin-1 (ET), insulin-like growth factor-1 (IGF-1), integrins, and Wnts [86]. Therefore, miRNA regulation of TGF-β and CTGF signaling is of paramount interest. Up-to-date, 10 miRNAs showed consistent experimental evidence of being TGF-β/CTGF signaling regulators. Six of them were dysregulated in more than one fibrotic disorder (see Table 3).
miR-17-92 cluster. miR-17-92 is a proto-oncogenic cluster (also called oncomir-1) consisting of six miRNAs. Very recently, miR-18a and miR-19a/b from this cluster have been shown to regulate CTGF and thrombospondin-1 (TSP-1) in the context of liver and cardiac fibrosis [60, 61]. In a previous report, CTGF and TSP-1 were validated as direct miR-18a/miR-19a/b targets in cancer [87]. Kodama et al. [60] reported down-regulation of the miR-17-92 cluster in liver fibrosis in a p53-dependent manner and an associated up-regulation of CTGF. Similarly, van Almen et al. [61] showed down-regulation of the miR-17-92 cluster and spontaneous upregulation of CTGF and TSP-1 in a mouse model of age-related heart failure, which is characterized by widespread interstitial EMC accumulation. Down-regulation of the miR-17-92 cluster paralleled by CTGF and TSP-1 gene up-regulation was confirmed in human idiopathic cardiomyopathy specimens. Transfection experiments in aged neonatal rat cardiomyocytes were used to confirm CTGF and TSP-1 modulation by miR-18a and 19a/b. Down-regulation of the miR-17-92 cluster has also been reported in human IPF lung tissue [48].
miR-133, miR-30 and miR-590. Two other major regulators of CTGF expression in cardiac fibrosis are miR-133 and miR-30. miR-133 is a cardiac specific miRNA and its expression is limited to cardiomyocytes, while miR-30 is expressed in both cardiac fibroblasts and cardiomyocytes as well as in other tissues. Duisters et al. [52] reported miR-133- and miR-30c-mediated repression of CTGF in two animal models of overloaded heart disease: a rat renin-dependent hypertensive failing heart model (Ren2 rats) and a mouse mechanical stress-induced model (mice undergone transverse aortic constriction). They first showed down-regulation of the two miRNA families and up-regulation of CTGF in vivo, then confirmed the direct interaction of miR-133 and miR-30c with CTGF mRNA in vitro using cultured cardiomyocytes and cardiac fibroblasts, and finally proved the functional effects of this interaction on collagen synthesis. Interestingly, the authors also showed that miR-133 and miR-30c do not exert additive effects on CTGF regulation, which might be explained by the overlapping seed regions of these miRs in the 3’UTR of the CTGF gene. Similar down-regulation of miR-133 and miR-30c and increased levels of CTGF were observed in human hypertrophic heart.
The importance of miR-133 down-regulation in cardiac fibrosis is further supported by the findings of Shan and colleagues [53], who investigated the expression of miR-133 in the heart of smokers with chronic atrial fibrillation (AF), and in a canine model of nicotine-induced AF. Fibrotic structural remodeling was strongly induced by nicotine in both human and canine heart tissue. This was associated with an increase in TGF-β1 and the TGF-β pathway downstream targets CTGF and collagens both in vivo and in vitro. By in silico analysis, the authors selected miR-133 and miR-590 as potential regulators of TGF-β1 and TGF-βRII, respectively. miR-133 and miR-590 were strongly down-regulated in dog atrial fibroblasts treated with nicotine, and the direct interaction of the two miRNAs with their predicted targets could be confirmed by reporter gene assay. miR-30c down-regulation has also been reported in human IPF lung tissue [48].
miR-200. Most data link the five miR-200 family members to EMT [54, 66, 88], thus indicating an indirect role in fibrogenesis. Recent data from Wang et al. [54] showed in addition that the miR-200 family contributes directly to fibrosis by regulation of TGF-β2. Expression of miR-141 and miR-200a has been investigated in mouse models of early (ApoE knockout mice injected with streptozotocin) and advanced (C57BL6 mice treated with adenine) diabetic nephropathy, as well as in rat proximal tubular epithelial cells (NRK52E). Both miRNAs were down-regulated in mice, but only miR-200a was sensitive to TGF-β stimulation in NRK52E cells. At the same time, TGF-β stimulation induced transformation of NRK52E cells towards a mesenchymal phenotype as indicated by reduced E-cadherin expression and increased vimentin and α-SMA expression along with enhanced ECM synthesis. In turn, forced up-regulation of both miR-141 and miR-200a in NRK52E cells reduced TGF-β2, Smad3 activity and ECM proteins, and also prevented EMT, suggesting a miR-200a/TGF-β feedback loop in the pathogenesis of diabetic CKD. In addition, reporter gene assays confirmed TGF-β2 as a direct target of miR-141/miR-200a.
miR-21. Like the miR-17-92 cluster, miR-21 is traditionally regarded as an oncomir, and due to its ubiquitous expression it is one of the most widely investigated miRs [89]. Evidence for an important role of miR-21 in fibrosis comes from studies on heart, lung and kidney [68-70]. Thum and colleagues [69] described miR-21 up-regulation in three mouse models of heart failure: β1-adrenergic receptor transgenic mice, C57BL/6 mice undergone transverse aortic constriction, and C57BL/6 mice treated with isopropanol. In different cell types isolated from failing hearts, miR-21 was predominantly up-regulated in cardiac fibroblasts, and mediated protection from apoptosis. Anti-miR-21 treatment and extracellular signal-regulated kinase/mitogen activated protein kinase (ERK/MAPK) blockade induced apoptosis to a similar extent, suggesting a possible regulation of ERK/MAPK signaling by miR-21. Accordingly, overexpression of miR-21 in both mouse and rat fibroblasts led to increased expression and activation of ERK/MAPK proteins. The authors identified sprouty1, a negative regulator of ERK/MAPK, as the direct target of miR-21 mediating the effects on this pathway. Nevertheless, more recently, Patrick et al. [90] demonstrated in mir21-/- mice, which were exposed to different stimuli resulting in cardiac overload, that miR-21 contributes to, but is not essential for ERK/MAPK activation in stress-dependent cardiac remodeling.
Liu et al. [68] confirmed the pro-fibrotic activity of miR-21 in human IPF and murine bleomycin-induced lung fibrosis. Like in heart failure, miR-21 up-regulation was proportional to the severity of lung fibrosis in the animal model. TGF-β1 expression showed the same trend. Therefore, the authors elucidated the causal relation between miR-21 and TGF-β1, showing that miR-21 expression was reduced in TGFβRII dominant negative mice injected with bleomycin. Consistently, TGF-β1 treatment of cultured pulmonary fibroblasts induced miR-21 up-regulation and, in turn, miR-21 overexpression induced TGF-β1 expression, Smad2 activation, and ECM synthesis. These effects were mediated by direct repression of Smad7, the inhibitory Smad which counter-regulates Smad2/3 activation. Finally, administration of miR-21 antisense oligonucleotides in vivo attenuated bleomycin-induced lung fibrosis.
Studies on the role of miR-21 in renal fibrosis further elucidated the interaction of this miRNA with TGF-β/Smad signaling [70]. In these experiments, TGF-β-mediated upregulation of miR-21 in tubular epithelial cells was prevented by knockdown of Smad3, but not by knockdown of Smad2. Consistently, Smad3 deficient mice, but not Smad2 deficient mice, exposed to kidney injury by unilateral urethral obstruction were protected from miR-21 up-regulation and fibrosis.
miR-216, miR-217 and miR-192. miR-216 and miR-217 are two clustered miRNAs encoded in the second intron of the RP23 gene. They showed pro-fibrotic effects in murine mesangial cells (MCs) [71]. Kato et al. reported upregulation of miR-216/217 in mouse MCs following TGF-β treatment, with consequent amplification of TGF-β signaling. miR-216 and miR-217 directly target phosphatase and tensin homolog (PTEN), which inhibits the phosphoinositide-3-kinase pathway (PI3K), thereby preventing activation of Akt. Thus, down-regulation of PTEN de-represses Akt signaling in MCs leading to prolonged cell survival, hypertrophy and increased collagen synthesis. In addition, since the promoter region of the RP23 gene contains multiple Smad binding elements and E-boxes, the authors could demonstrate that the expression of miR-216 and miR-217 in MCs was induced by TGF-β and zinc finger E-box-binding homebox (Zeb) 1 and 2. In turn, Zeb 1 and 2 are regulated by miR-192 [62].
MicroRNA REGULATION OF ECM PROTEINS
miR-29. miR-29 is the best characterized direct regulator of ECM synthesis. Its involvement in fibrosis as an anti-fibrotic miRNA has been reported in SSc as well as in other major fibrotic disorders. The miR-29 family includes three members: miR-29a/b/c. We could show that all three miRNAs are down-regulated in human SSc dermal fibroblasts and SSc skin [47]. miR-29a showed the strongest down-regulation in tissues and cells, and the most consistent functional effects on the expression of collagen type I and type III. In addition, COL3A1 was validated as a direct target of miR-29a. We also demonstrated that PDGF-B and TGF-β down-regulate miR-29a, and that down-regulation of miR-29a leads to further upregulation of PDGF-B and TGF-β. This creates a positive feed-back loop among profibrotic mechanisms leading to the uncontrolled release of extracellular matrix proteins in SSc. Results were confirmed in a bleomycin-induced mouse model of skin fibrosis, in which the tyrosine-kinase inhibitor imatinib mesylate was able to restore miR-29a levels.
In murine and human hearts, down-regulation of the miR-29 family following myocardial infarction has been reported [74]. Van Rooji et al. showed that cardiac fibroblasts, but not cardiomyocytes, are responsible for the effects of miR-29 on ECM synthesis in myocardial infarction. They hypothesized that miR-29 down-regulation is mediated by TGF-β following cardiac injury, and that this axis is controlled by the parallel induction of brain natriuretic peptide (BNP), which causes repression of TGF-β. COL1A1, COL1A2, COL3A1, FBN1, and ELN1 genes were validated as direct miR-29 targets. In addition, miR-29b knockdown with cholesterol-conjugated antagomirs in C57BL/6 male mice induced COL1A1, COL1A2, and COL3A1 mRNA overexpression in cardiac tissue.
A clear anti-fibrotic function of the miR-29 family was also shown in hypertensive kidney disease [76]. Liu et al. found that miR-29b was overexpressed in a mouse model, which is protected against salt-induced hypertensive renal medullary fibrosis (SS-13BN) after high-salt diet. Silencing of miR-29b in the kidney of these animals caused up-regulation of a large number of ECM and ECM-modulating genes, subsequently validated as direct targets of miR-29b by reporter gene assay (COL1A1, COL3A1, COL4A1, COL5A1, COL5A2, COL5A3, COL7A1, COL8A1, MMP2, and ITGB1). As reported by Du et al. [77], miR-29a down-regulation can also be induced in vitro in human proximal tubular epithelial cells (HK-2 cells) by high glucose and TGF-β stimulation, with similar effects on the expression of multiple collagen genes, suggesting a role for miR-29 also in diabetic nephropathy.
Regarding lung fibrosis, down-regulation of miR-29 has been reported by Cushing et al. [75] in bleomycin-treated mice, which was paralleled by an increase in COL3A1 and COL4A1 expression. In addition, the same authors investigated the effects of silencing of miR-29 family members in human fetal lung fibroblasts. They identified new direct targets, among them both structural ECM genes and enzymes involved in tissue remodeling, namely NID1, ITGA11, ADAMTS9, and ADAMTS12. In lungs as well as in the skin, heart and kidney, TGF-β was able to reduce miR-29 levels.
Lastly, miR-29 down-regulation (miR-29c) has been reported in a mouse model of non-alcoholic steatohepatitis [59], although functional investigations of miR-29 in the liver are still lacking.
Other miRNAs. Additional miRNAs have been shown to regulate ECM synthesis in the kidney: miR-377 and miR-449. Wang et al. [81] showed pro-fibrotic effects of miR-377 in human mesangial cells following high glucose and TGF-β administration and in animal models of type I diabetes. These effects were mediated by an indirect upregulation of fibronectin via targeting the fibronectin inhibitors p21-activated kinase and superoxide dismutase. Muth et al. [83] demonstrated that the expression of miR-449a/b was reduced in human fibroblasts cultured under hypoxic conditions and in human kidney transplants displaying chronic allograft nephropathy. miR-449 down-regulation induced Serpine-1 (PAI-1) expression. Serpine-1 inhibits tissue-type (tPA) and urokinase-type plasminogen activator (uPA), which mediate proteolysis of ECM proteins and activation of latent metalloproteinases (MMPs). Thus, the authors hypothesized that miR-449 down-regulation promotes ECM accumulation in hypoxic fibroblasts and chronic post-transplant allograft remodeling in the kidney.
CONCLUSIONS AND PERSPECTIVES
Our review outlines the potentially important role of miRNAs in fibrosis. With some exceptions, most of the miRNAs reported in this review are ubiquitously expressed, indicating that the majority of pro- or anti-fibrotic miRNAs might be common regulators of fibrosis in different organs and disease settings. In addition, several key pro-fibrotic molecules are regulated by multiple miRNAs [51-54, 60, 61, 68-71].
The partially redundant interaction between miRNAs and their targets might hamper the use of miRNA mimics or inhibitors as potential therapeutic tools. However, data on the use of antagomirs in animal models show promising results [68, 74, 90]. It is important for the development of targeted therapies that miRNAs exert rather mild to moderate effects on single molecules, but that these effects are additive when multiple molecules of a certain pathophysiological pathway are regulated. Therefore, miRNAs are of particular interest for targeted therapies in fibrosis, when they regulate multiple rate limiting molecules of profibrotic pathways. In addition, since differentially expressed miRNAs are often disease specific and do not play major roles under normal conditions in adult tissue or in quiescent cells, targeting of an aberrantly expressed miRNA is expected to be well tolerated [91]. Indeed, many miRNA knockout animal models do not display a specific phenotype unless they are challenged with a stimulus causing cellular stress in target organs [69]. However, the miRNA machinery in fibrosis is still far from being fully elucidated, although research in this field in the last years has been considerably fostered.
ACKNOWLEDGEMENT
Supported by a grant from the EULAR ODP program.
CONFLICT OF INTEREST
O. Distler had consultancy relationship and/or has received research funding from Actelion, Pfizer, Ergonex, BMS, Sanofi-Aventis, United BioSource Corporation, medac, Biovitrium, Boehringer Ingelheim Pharma, Novartis, 4 D Science, Bayer and Active Biotec in the area of potential treatments of scleroderma and its complications, he is holding a patent on miR-29 for the treatment of scleroderma.

