All published articles of this journal are available on ScienceDirect.

Cellular Senescence is a Common Characteristic Shared by Preneoplasic and Osteo-Arthritic Tissue
Authors Info & Affiliations
Abstract
Objective:
This study aims at highlighting the common signature between cartilaginous tissue in osteoarthritis (OA) and preneoplasic tissues preceding neoplasia and tumour formation and, second, focusing on the molecular mechanisms at the aetiology of both pathologies.
Results:
Because age is the highest risk factor common for both OA and cancer development, it is tempting to compare the molecular mechanisms occurring at the onset of OA and preneoplasic lesions. Indeed, cellular senescence seems to be a common characteristic. Cellular senescence represents a natural barrier to suppress the unscheduled proliferation of damaged cells acting as a strong tumour suppressor pathway and in OA, it also occurs prematurely in chondrocytes. In this study, we review a number of molecular factors associated with the senescent phenotype.
Conclusion:
Whereas accumulation of senescent cells in preneoplasic-like lesions leads to tissue degeneration and potentially tumour development; in OA, senescent cells accumulate in a slowly proliferative tissue. This is likely contributing at reducing the risk of cell transformation.
INTRODUCTION
Osteoarthritis (OA) is a chronic degenerative disease characterized by progressive cartilage erosion leading to pain. At present, there is no efficient treatment to cure or even delay the progression of this most common disability-associated disease. The aetiology of OA is still unknown although clinical studies have shown that numerous factors are strong determinants. Among them, can be included genetic predisposition, obesity and primarily, age [1]. Remarkably, all of these factors are also associated with the prevalence of other age-related human diseases such as atherosclerosis [2] and preneoplasic lesions [3]. Recent works have focussed on the molecular events that govern the genesis of these lesions accumulating with age in numerous tissues [4, 5]. Preneoplasic lesions are benign. Nevertheless these cells may acquire, with time, new properties, such as genetic instability inducing in fine cellular transformation leading to neoplasia and eventually tumour formation. In summary, preneoplasic lesions are seen as natural barriers preventing tumour expansion and malignancy. Analysis of these recent studies reveals the strikingly high number of common characteristics between preneoplasic tissues and OA cartilage. Even if these common characteristics of cellular senescence in both pathologies may be merely co-incidental, they have to be highlighted in the prospect of using new therapeutic intervention in OA. Thus, the aim of this review is first, to highlight their common signatures and second, to focus on the molecular mechanisms shared by both pathologies.
ARTICULAR CARTILAGE BIOLOGY
The role of articular cartilage is to transmit forces across diarhodial joints and maintain a friction-free surface at the epiphyseal ends of long bones to support limb movement. Articular cartilage is organised in various layers and contains one cell component, the chondrocyte which contributes by its anabolic function to the production and/or maintenance of the extracellular matrix (ECM) of the tissue [6]. The ECM is composed of a network of collagen fibres, primarily type II collagen and secondarily, type IX and XI collagens that give the tissue its shape, strength and tensile force. It also contains a highly hydrated gel of large proteoglycans, aggrecan being the most prominent, and glycoproteins which give resistance to compression [7]. Once the cartilage is formed in the adult, the turn-over of ECM components is very low.
OA PHYSIOPATHOLOGY
Among the numerous diseases which affect cartilage, OA is the most prevalent one. A number of studies have described the progression of this common disease [8]. The first detectable changes during OA by clinical examination are softening, fibrillation and ulceration of the cartilage. At latest stages of the disease, appearance of chondrophytes from neodifferentiated periosteal cells which eventually turn after calcification into osteophytes are present on the joint edge [9]. These osteophytes, as a sort of extension of the articular surface, are responsible of the increasing pain. Finally, subchondral bone remodelling is proposed to be one driving force that accelerates cartilage [10].
Chondrocytes are the central players in the changes that occur during primary or secondary OA [8]. Anabolic function of chondrocytes is decreasing following pathology progression. This loss of function is mainly associated with a premature cellular senescence of chondrocytes [11]. Indeed, OA chondrocytes express a panel of senescence markers including acidic β-galactosidase activity [12], increase in reactive oxygen species (ROS), telomere attrition [13] and p53/p16ink4 accumulation [11]. Although the subject of debate, apoptotic chondrocytes have also been detected by tunnel assay in OA cartilage [14, 15]. Furthermore, an increased number of chondrocytes with altered phenotype and chondrocyte progenitors have also been found in OA cartilage [8, 16]. Finally, an increased number of hypertrophic chondrocytes, characterised by the expression of specific markers, such as collagen type X, alkaline phosphatase or runx2, is frequently observed [17]. In summary, OA is a multi-phenotypic complex disease where articular chondrocytes exhibit characteristics of stress-induced senescence with altered phenotypes and impaired normal anabolic capacities [11].
NEOPLASIA PHYSIOPATHOLOGY
Preneoplasic and neoplasic lesions are common. Both types of lesions are located within a high proliferative tissue and they increase with age. They are the result of abnormal cell proliferation that exceeds the surrounding normal tissue. Physiologically, preneoplasia preceed neoplasia and both stages of lesion progression are hardly distinguishable. Neoplasia can remain benign or degenerate into tumour. Preneoplasia is induced in vivo following viral infection, recurrent genotoxic insults such as ultra-violet (UV), metabolic oxidative stress or genetic predisposition [18]. Genetic and molecular events that initiate this dysplasia have been broadly documented for the last 15 years. Indeed, these abnormal cells harbour an apparent attempt to hyperproliferate but rather are kept confined by a permanent cell cycle arrest associated with a senescent phenotype and impaired secretor activities [5, 19-21].
SIMILARITIES BETWEEN OA- AND PRENEOPLASIA/ NEOPLASIA-ASSOCIATED MOLECULAR FACTORS
Cells are naturally exposed to various types of stress that lead to genomic assault during lifespan. Thus, cell-intrinsic mechanisms have evolved to limit the expansion of genetically altered cells. Cellular senescence is one of the natural barriers developed by multicellular organisms to suppress the unscheduled proliferation of damaged cells [19, 22, 23]. Cellular senescence is therefore a strong tumour suppressor pathway that is activated in benign preneoplasic-like lesions [5, 19, 21]. Remarkably, cellular senescence also occurs prematurely in OA chondrocytes. It is therefore tempting to make analogies between OA and preneoplasia associated characteristics (Fig. 1).
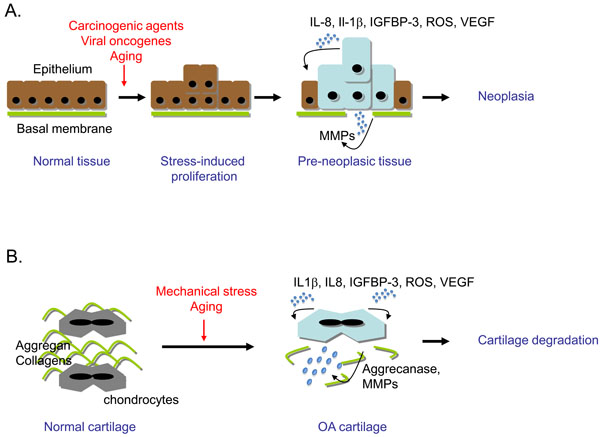
Cellular senescence is a common characteristic of preneoplasic tissue and cartilage in early osteoarthritis (OA). (A) Neoplasia genesis. Upon environmental stress (carcinogens, oncogenes, genotoxics), in highly proliferative tissues such as epithelium, some cells will respond by abortive proliferation and emergence of senescent cells. If one cell overcomes the senescent process by inactivation of the DNA damage response (DDR), cell proliferation will lead to neoplasia and eventually tumour. (B) OA genesis. Upon environmental stress (mechanical stress, aging factors), OA chondrocytes become senescent and display an imbalance between anabolic and catabolic activity in favour of catabolism, leading to cartilage degradation. In both tissues, common senescence-associated markers are expressed: activated DDR and secretion of reactive oxygen species (ROS), interleukin (IL)-1β, IL-8, insulin growth factor binding protein (IGFBP)-3, vascular endothelial growth factor (VEGF) and metalloproteinases (MMPs).
ROS and Associated Cellular Senescence
Oxidative damage and the chronic production of endogenous ROS have been associated with aging in various human tissues and in animal models [24]. Articular chondrocytes actively produce ROS under mechanical stress or after inflammatory treatment [25, 26] and ROS secretion is believed to be one major mediator of OA-associated physiopathology [27]. Indeed, in vitro treatment of chondrocytes with oxygen peroxide (H2O2) used as source of ROS, resulted in p53/p21Waf1/Cip1, p16ink4a accumulation and telomere attrition which are both hallmarks of cellular senescence [12, 28, 29]. Furthermore, it has been reported that high level of ROS are produced by chondrocytes from OA patients [27, 30]. These molecular markers were also found in cartilage from patients with early OA suggesting their roles in the pathogenesis of the disease [11].
Interestingly, ROS have been proposed to play a pivotal role in the genesis of preneoplasic tissues [4]. Indeed, treatment of cells with ROS or transduction of primary epithelial cells or fibroblasts with oncogenes inducing preneoplasia in vivo led to the rapid increase of intracellular ROS production [31]. ROS participated to the DNA damage accumulation and thereby to the activation of the DNA damage response (DDR) [4]. The DDR is the signalling pathway orchestrated by ATM/ATR kinases (Ataxia telangiectasia mutated and ATM-Rad3 related) transducing toward two checkpoint kinases namely Chk1/Chk2. The DDR converges to p53 and induces senescence-dependent cell cycle arrest characteristic of preneoplasic cells. Therefore, DDR activation induced by ROS accumulation is now believed to be the hallmark of in vivo senescent tissues [22, 32].
Inflammation and Pro-Inflammatory Cytokines
Although OA is classified as a non inflammatory disease, pro-inflammatory cytokines such as IL-1β, TNF-α or IL-8, have been reported to participate in OA physiopathology. Indeed, IL-1β and IL-8 were detected both in the synovial fluid and in cartilage from OA patients [33, 34]. Depending on cell culture conditions, IL-1β-treated chondrocytes underwent either dedifferentiation or telomere dependent-cellular senescence [29]. IL-8 has been proposed to induce chondrocyte hypertrophy and osteophyte formation which are observed in OA cartilage. Indeed, a time and dose-dependent increase in the expression level of specific hypertrophic markers was observed in OA chondrocytes treated with IL-8 [35]. Nevertheless, the role of this cytokine remains unclear.
For decades, IL-1β and IL-8 are also associated with pre-malignant and malignant tumour lesions [36]. These inflammatory cytokines together with IL-6 were recently shown to play a crucial role in the premature cellular senescence found in preneoplasic lesions [36-38]. Indeed, when over-expressed in primary fibroblasts, IL-8 and one of its known receptor, CXCR2 were sufficient to induce premature senescence (acidic β-galactosidase staining and DDR activation) [38]. Furthermore, IL-8/CXCR2 tandem is transcriptionally induced by the DDR and participates to the senescence-dependent cell cycle arrest by producing more ROS [39]. Therefore, these signalling pathways including IL-8, IL-6 or Il-1β were postulated to be tumour suppressors with pro-aging properties and accordingly found activated in benign lesions and inactivated during tumour progression [19, 38, 40, 41].
IGFPB Family Members
Differential analysis of gene and protein expression in OA versus healthy age-matched chondrocytes has revealed the over-expression of members of the IGFBP family such as insulin growth factor binding protein (IGFBP)-3 and -7 [42, 43]. IGFBP family members are pro-aging secreted factors known to modulate the insulin signalling pathway (for review, see [20]). Indeed, insulin pathway is central to aging and senescent mechanisms in modulating ROS production and cell survival [20, 44]. IGFBP-2/3 and -7 have also been shown to accumulate in preneoplasic-type lesions contributing to cellular senescence of these cells [3, 19, 45].
Vascularisation and VEGF Production
Another characteristic of OA cartilage is an increased expression level of vascular epithelial growth factor (VEGF). This growth factor is secreted by hypertrophic chondrocytes and contributes to vascularisation and attraction of cartilage-resorbing cells during endochondral ossification in long bone development. In OA patients, VEGF production was shown to participate in the formation of osteophytes at the edges of articular cartilage [46]. However, the signalling pathways which control transcriptional induction of VEGF in OA chondrocytes are still being dissected. On the contrary, the transcriptional regulation and role of VEGF were broadly documented in early tumour development. Indeed, fibroblasts genetically modified to express oncogenes such as v-Ras or senescent fibroblasts were shown to secrete high levels of VEGF that can stimulate the proliferation of premalignant epithelial cells in culture and facilitate their conversion to malignant tambours in vivo [47]. This finding is believed to control the neo-vascularisation of neoplasic-like tissues and tumour cell survival to hypoxia-induced apoptosis.
Secretion of ECM Proteinases
Characteristic changes in early OA are similar to those seen with aging in cartilage from older, healthy subjects, with collagen damage and collagenases concentrated closer to the articular surface. Indeed, ECM degradation in OA was characterized by the secretion of metalloproteinases and aggrecanases such as MMP-1, -3, -8, -13 and a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS)-5, respectively [48, 49]. Genetic inactivation of ADAMTS-5 prevented cartilage degeneration in OA-induced mice models [50, 51]. Recent studies revealed that senescent cells from preneoplasic tissues also promoted ECM degradation via the secretion of metalloproteinases such as MMP-3 [52]. The presence of metalloproteinases within preneoplasic lesions is believed to create a specific substratum which thereby facilitates the potential changes in cellular morphology required for tumour progression and metastasis [3].
CONCLUSIONS AND PERSPECTIVES
Cellular senescence seems to be a common process developed by OA chondrocytes and pre-tumour cells that may be at the genesis of both pathologies. Prevalence of these diseases increases with age and among the senescence-associated markers, ROS accumulation plays a central role. ROS production induces DDR-dependent premature senescence in preneoplasic cells and in OA chondrocytes contributing to the loss of their anabolic functions.
A number of studies now suggest that senescent cells secrete specific factors that set the stage for a cross-talk between senescent cells and their environment [20]. Such signals would be integrated into a coordinated response that can cause other cell components to prematurely senesce thereby establishing a pro-aging loop [20]. The consequence of the pro-aging loop is the degradation of the ECM, degeneration of the targeted tissue and production of survival factors such as VEGF [37]. Indeed, OA chondrocytes and preneoplasic-associated senescent cells both secrete VEGF but in pre-tumour tissues, VEGF helps tissue growth and survival by promoting new blood vessel formation [47]. In contrast, in the cartilage, the increase in vascularisation participates to the aberrant ossification of cartilage leading to osteophyte formation [46]. This phenomenon would favour subchondral bone pressure in OA patients.
Two exciting points of view emerge from all the similarities reported here between both diseases. First, as we mentioned, the aetiology seems to be similar. In proliferative tissue such as epithelium, the accumulation of senescent cells within preneoplasic-like lesions harbouring specific secretary properties will lead to tissue degeneration and may induce tumour development [5, 20]. By chance, accumulation of premature senescent chondrocytes in OA lesions occurs in a slowly proliferative tissue, dramatically reducing the risk of cell transformation yet promoting tissue degeneration. Second, research on OA physiopathology should take into account the literature on the deciphering of the neoplasia-associated molecular mechanisms and the prevention of tumour formation. Indeed, according to the numerous similarities between preneoplasic and OA lesions, it would be questionable whether the battery of pharmaceutical compounds available for anti-cancer treatments such as anti-VEGF treatments could be tested by clinicians as potential OA treatments in order to delay or prevent the progression of this disease.
ACKNOWLEDGEMENTS
Work in the laboratory Inserm U844 is supported by the Inserm Institute, the University of Montpellier I and grants from the “Fondation Pour la Recherche Médicale”.

