All published articles of this journal are available on ScienceDirect.

The First Reported Case of Juvenile Dermatomyositis in an Adolescent with Down Syndrome and the Clinical Considerations for Therapy
Abstract
Background:
Down Syndrome (DS) is one of the most common birth conditions in the United States of America, with approximately 5300 births annually, resulting in an estimated birth prevalence of 12.6 per 10,000 live births and a population prevalence in the USA since 2010 of 6.7 per 10,000 inhabitants. Children with DS have complex medical challenges that present due to changes in their immune system that results in increased rates of infection, malignancy, and autoimmune disease. Juvenile Dermatomyositis (JDM) is a rare, autoimmune disease, and the most common inflammatory myopathy of childhood. Reports suggest an increased incidence of arthritis in children with DS, but there have been no reports of JDM in children with DS. Additionally, those with DS pose unique challenges with an increase in adverse effects and ineffectiveness of immunosuppressive therapy.
Case :
We describe the first case of an adolescent female with DS who developed JDM with a positive anti-p155/140 antibody and characteristic clinical phenotype. We discuss increased awareness of autoimmune disease in adolescents with DS and clinical considerations for therapy with immunosuppression.
Conclusion:
Adolescents with DS are at increased risk for autoimmune disease, including JDM. Awareness, early recognition of the signs and symptoms of autoimmune disease in those with DS is important, so appropriate therapy can be implemented.
1. INTRODUCTION
Down Syndrome (DS) is one of the most common birth conditions in the United States of America, with approximately 5300 births annually, resulting in an estimated birth prevalence of 12.6 per 10,000 live births [1] and a population prevalence in the USA since 2010 of 6.7 per 10,000 inhabitants [2]. Children with DS have complex medical challenges that are present due to changes in their immune system resulting in increased rates of infection, malignancy, and autoimmune diseases such as Celiac and thyroid disease [3]. More recent reports suggest an increased incidence of arthritis in children with DS [4, 5], but there have been no reports of Juvenile Der Matomyositis (JDM) in children with DS. JDM is a rare, auto immune disease and the most common inflammatory myopathy of childhood [6].
This report focuses on a 16-year-old female who was presented to the emergency department with a rash and is the first report of juvenile dermatomyositis occurring in a child with Down syndrome.
2. THE CASE
A 16-year-old African American female with Down syndrome was presented to our institution for worsening skin rash and hair loss. Prior to the presentation, she was noted to have a history of milk allergy and eczema, but over a few months’ time, her skin had progressively worsened with erythema, hyperpigmentation and scaling on her head, face, neck, arms, and legs. The family was concerned that she had accidentally ingested milk. She also developed hair thinning that progressively worsened. The rash was intermittently pruritic, but not painful and was noted to be more erythematous with prolonged sun exposure. Additional symptoms included decreased appetite, and several lower extremity abrasions and contusions she had recently sustained from multiple falls. She was also noted to have less endurance in gym class at school over the past several months. Mother and patient denied any cough, dysphagia, chest pain, shortness of breath, dyspnea, abdominal pain, or joint pain. There was no significant family history, regular daily medications or new environmental exposures that could be identified. The family presented to the Emergency Department (ED) due to worsening of the rash. In the ED, dermatology was consulted, and she was started on more topical steroids, an antihistamine and laboratory tests were obtained, which showed leukopenia (2.8 K/μL), neutropenia (1.05 K/μL), and anemia (11.2 gm/dL), with appropriate complete metabolic panel including normal aspartate aminotransferase (AST) and alanine aminotransferase (ALT), normal C-Reactive Protein (CRP), and normal erythrocyte sedimentation rate (ESR; 18 mm/hr; normal is less than 20 mm/hr). She was discharged from the ED to follow-up with dermatology, and subsequently referred to rheumatology for further evaluation for systemic disease.
Upon presentation to the rheumatology clinic, it was noted that she had muscle weakness with inability to do a sit-up, difficulty rolling from supine to prone position, and difficulty standing from a seated position on the floor. She also had limitation, tenderness, and swelling of bilateral wrists, left elbow, right hip, and right knee consistent with arthritis. Pes planus and ligament laxity were noted. She had erythematous papules on the metacarpophalangeal and proximal interphalangeal joints bilaterally, consistent with Gottron papules (Fig. 1), along with malar rash and erythema on her upper chest and back consistent with a shawl sign (Fig. 2). She also had capillary nailfold telangiectasias. Additional laboratory evaluation revealed normal peripheral smear, positive ANA (1:640), elevated IgG (1990 mg/dL; normal range 613 - 1295 mg/dL), elevated von Willebrand Factor antigen (244; normal range 52-175) with normal Creatine Kinase (CK), aldolase, Lactate Dehydrogenase (LDH), ESR, CRP, and complement levels. She was also found to have negative rheumatoid factor, negative double-stranded DNA antibody, negative extractable nuclear antigen antibody panel and negative anti-phospholipid antibodies. A myositis antibody panel was obtained and was positive for p155/140 antibody. The myositis antibody panel is a comprehensive profile that evaluates several myositis-specific and myositis-associated antibodies. An MRI of the thighs was ordered to assess for myositis and showed diffuse myositis of the pelvic and thigh musculature, bilateral tendinopathy involving the gluteus medius and vastus lateralis tendon insertions. A CT scan of the chest, abdomen, and pelvis was done to evaluate for malignancy, all of which was negative. The constellation of muscle weakness, skin findings (Gottron papules, malar rash, shawl sign, capillary nailfold changes), and arthritis concerned Juvenile Dermatomyositis (JDM). One of the challenges of this case was the minimal serologic abnormalities with normal levels of CRP, ESR, AST, ALT, LDH, CK and aldolase; however, there was clear clinical evidence of weakness, pathognomonic skin rashes, with positive MRI findings consistent with proximal muscle myositis, positive myositis antibody (p155/140) and negative malignancy evaluation, which all supported the diagnosis of JDM.
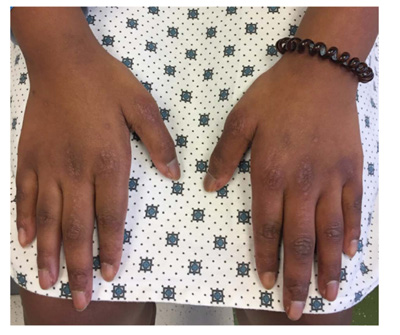
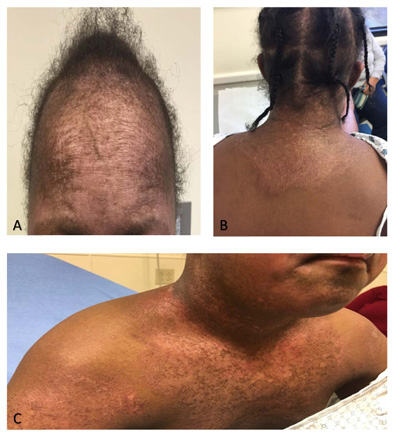
Her initial treatment plan followed consensus treatment protocols for JDM(7) and included: intravenous (IV) immunoglobulin (2 g/kg with a max of 70 grams) infusions every 2 weeks for three doses and then monthly thereafter, IV methylprednisolone (1000mg) daily for three days then taper to weekly along with 20 mg daily oral prednisone and twice daily cyclosporine (100mg). However, after discussion with the family, the IV therapies were refused, and the family agreed to start oral prednisone (30 mg daily) and cyclosporine (100 mg twice daily). She responded well to therapy with decrease in arthritis, myositis, and improvement of skin changes. She was no longer falling, had improved endurance in gym class, and had less erythema with her rash. She started to grow hair in areas that previously were thin. After two months of therapy, she had no evidence of arthritis or myositis on exam, and her rash continued to improve but was still present. Her rash is still present on her extremities, hands, chest, face, and back but improved with hyperpigmentation and little to no erythema. Her current therapy includes 5 mg of oral prednisone and 100 mg twice daily cyclosporine.
3. RESULTS AND DISCUSSION
This is the first report of JDM in a child with DS. Children with DS have complex medical challenges that present due to changes in their immune system. Care for children with DS can be especially challenging as many children present with joint laxity and hypotonia, which can lead to delay in ambulation or intermittent problems with ambulation and endurance. This is likely the reason the patient’s falling and decrease in endurance were not initially concerning. Additionally, many of the initial laboratory tests were within normal limits, which added to the complexity. Heightened awareness is required when caring for children with DS because they have musculoskeletal disorders that may conceal the underlying problem, and with an increased incidence of autoimmune diseases they can be easily missed or unintentionally dismissed.
The incidence rate of JDM in the United States is 2.3 cases per million people per year for children with a mean age of onset of 6.7 years for boys and 7.3 years for girls [8] It is a systemic vasculopathy characterized by symmetric, proximal muscle weakness, pathognomonic skin rashes that include Gottron papules over extensor joint surfaces, heliotrope rash over the eyelids, malar rash and nailfold capillary changes, and raised serum concentrations of muscle enzymes [6]. The diagnosis of JDM is made through a constellation of clinical and laboratory tests, as applied in the 1975 criteria by Bohan and Peter [6]. The criteria for diagnosis are symmetrical proximal muscle weakness and presence of characteristic rash, increased muscle enzymes, characteristic myopathic electromyography (EMG) findings, and characteristic pathological changes on muscle biopsy. MRI is often used in place of EMG and muscle biopsy as it is less invasion and very sensitive method to assess muscle inflammation. Additional skin findings may include inflammatory skin changes in the shawl area on the upper anterior chest and upper posterior back, which can be indicative of both acute and chronic changes, and scalp involvement resulting in alopecia. The comprehensive myositis profile used in this case screens for myositis-specific antibodies, which help define subsets of JDM, and myositis-associated antibodies, which helps to identify symptoms of other connective tissues diseases that may overlap with JDM. Antibody testing in JDM has helped define phenotype and prognosis of specific subsets of JDM. The three most common myositis-specific antibodies in children are anti-P155/140, anti-MJ (NXP-2), and anti-MDA-5. Anti-P155/140 is more associated with severe cutaneous disease, photosensitive rash, lipodystrophy and chronic disease course [7, 8], which is consistent with how our patient presented. Rarer myositis-specific antibodies that are associated with the antisynthetase syndrome (myositis, interstitial lung disease, mechanics hands, Raynaud’s phenomenon, arthritis), includes Jo-1, PL7, PL12, OJ, EJ, KS, Zo and Ha and occurs in 5-8% of JDM patients. Myositis-associated antibodies, which include anti-PMScl, anti-U1RNP, and anti-Ro52 are found in 16-20% of patients with JDM and can occur in conjunction with myositis-specific antibodies [8].
Most data for treatment of JDM are from anecdotal, non-randomized case series, and consensus treatment protocols, which recommend a combination of IV methylprednisolone, oral prednisone, subcutaneous methotrexate, and IV immunoglobulin with a taper of the steroids as symptoms improve [7]. One randomized trial did show that the combination of prednisone with cyclosporine or methotrexate was better than prednisone alone and that prednisone with methotrexate had a better safety profile compared to cyclosporine [9]. However, children with DS have a well-documented clinical sensitivity to methotrexate for the treatment of leukemia and arthritis [10]. This had an impact on our treatment approach as individuals with DS have unique biochemical characteristics that can impact medication absorption, tolerance, and pharmacotherapy [11]. Due to this, we avoided methotrexate. Mycophenolate mofetil (reversible, selective, non-competitive inhibitor of inosine monophosphate) and tofacitinib (JAK inhibitor) were considered as there is emerging evidence supporting their use for JDM. Additionally, we wanted to use IV steroids and a lower dose of oral prednisone due to the side effect profile, issues with weight gain, obesity and comorbid issues associated with DS, and many rheumatologists will avoid steroids in those with DS due to the adverse effects [11-13]. Due to family preferences, we used higher dose of oral prednisone and many associated adverse effects were noted until weaned to a dose of less than 10 mg once daily.
CONCLUSION
In summary, we report the first case of an adolescent female with DS who developed JDM with positive anti-p155/140 and characteristic clinical phenotype. We discussed increased awareness for autoimmune disease in children with DS and implementation of appropriate therapy.
AUTHOR'S CONTRIBUTIONS
Dr. Jones and Chelsey Smith had full access to all of the case report information in the study and took full responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: N/A
Acquisition, analysis, and interpretation of data: Jones, Smith.
Drafting of the manuscript: Jones, Smith.
Critical revision of the manuscript for important intellectual content: Jones, Smith.
Statistical analysis: N/A
Obtained funding: N/A
Administrative, technical, or material support: N/A
Study supervision: N/A
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Informed consent was obtained from the patients when enrolled in the study.
STANDARDS OF REPORTING
CARE guidelines have been followed.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
The authors received no financial support for the research, authorship, and/or publication of this article.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ACKNOWLEDGEMENTS
Declared none.

