All published articles of this journal are available on ScienceDirect.

Vitamin D, Inflammation and Osteoporosis in Rheumatoid Arthritis
Abstract
Patients with Rheumatoid Arthritis (RA) commonly develop osteoporosis and fragility fractures. This fact cannot be explained only with the use of glucocorticoids, known to be detrimental for bone health. RA is characterized by a chronic inflammation caused by the continuous activation of innate and adaptive immunity with proinflammatory cytokines overproduction. This process is detrimental for several organs and physiological processes, including the impairment of bone remodeling. We will briefly review the pathogenesis of inflammation-related bone loss in RA, describing well-known and new molecular pathways and focusing on vitamin D and Parathyroid Hormone role.
1. OSTEOPOROSIS IN RHEUMATOID ARTHRITIS: EPIDEMIOLOGICAL EVIDENCES AND RISK FACTORS
Patients affected by inflammatory arthritis in general, and Rheumatoid Arthritis (RA) in particular, show an increased risk of generalized bone loss; this is the result of the coexistence of common risk factors, such as age, female gender, smoke and body mass index along with RA-related features [1].
Generalized osteoporosis, defined as a T-score -2.5 Standard Deviations (SD) in the hip and/or in lumbar spine as proposed by the World Health Organization (WHO), is highly prevalent among RA patients: in a large study on RA patients, a two-fold increase in osteoporosis rate was seen in 394 women, with respect to a control group; similarly a doubled rate of reduced Body Mass Density (BMD) was reported in men [2, 3]. In a more recent cohort of 304 RA consecutive patients, the prevalence of osteoporosis was 29.9%, almost two-fold of the one observed in 904 controls matched for age and sex [4].
Consistently, patients with RA are at higher risk for vertebral and non-vertebral fractures: according to a retrospective observational study on more than 47,000 RA patients, the incidence rate for osteoporotic fracture at typical sites was 9.6 per 1,000 person-years, 1.5 times higher than the rate observed in subjects not affected by RA [5]. Moreover, Weiss et al in 2010, reported a 2.9 OR for hip fractures and a 2.7 OR for vertebral fractures in patients with RA with respect to the general population [6].
It has been argued that the increased risk of fractures can be explained by the use of Glucocorticoids (GC), however this is only one of the RA-related factors explaining bone loss in inflammatory arthritis. Consistently, Van Staa et al. found out-patients with RA who were never exposed to GC, were in any case characterized by an increased Relative Risk (RR) (1.5, CI 95%: 1.2-1.9) for vertebral fractures up to 1.7 (CI 95%: 1.5-2.0) for hip fractures, suggesting that disease by itself is an important and independent risk factor for accelerated bone loss [7]. The increased risk of fractures in RA is, therefore, multifactorial and includes the general background risk (clinical risk factors, BMD) on top of RA-specific risk factors (Table 1) [8].
| General Risk Factors for Osteoporosis | RA Related Osteoporosis Risk Factors |
|---|---|
| Female sex | Steroids |
| Age | Systemic inflammation |
| Early Menopause | Osteoclasts activation |
| Amenorrhea | Osteoblasts inhibition |
| Asian or European Ethnicity | Disease activity |
| Low BMD | Disease duration |
| Steroids | High titer of rheumatoid factor |
| Smoke and Alcohol | High titer of ACPA |
| Vitamin D and Ca2+ deficiency | Inactivity due to joint pain |
| Prolonged immobilization | – |
| Low weight | – |
| Poor visual acuity | – |
Interestingly, bone loss occurs very early in the course of the disease, when other risk factors such as reduced mobilization are rarely present, testifying the relevance of the inflammatory state. In early arthritis, in fact, the bone loss is evidently related to the parameters of inflammation [9-12].
2. BONE LOSS AND SYSTEMIC INFLAMMATION
It is nowadays well known that systemic inflammation influences bone turnover, favoring bone resorption and dampening its formation. Proinflammatory mediators are involved in this process, as witnessed by their involvement in the pathogenesis of hormonal deficiency-induced bone loss. Nude female mice deficient in T cells, as well as TNF knock-out mice, are protected against bone loss induced by ovariectomy, while bone loss occurs if these mice are injected with T cells from wild-type mice [13]. Similarly, IL6-deficient mice are protected from oestrogen deficiency-induced bone loss [14]. Moreover, in postmenopausal women, anti-IL1 or anti-TNF therapy can limit the increase in bone resorption markers which occurs after discontinuation of oestrogen replacement therapy [15].
In the last years, new insights into the field of osteoimmunology expanded our knowledge, leading to a better comprehension of the biological systems involved in this process.
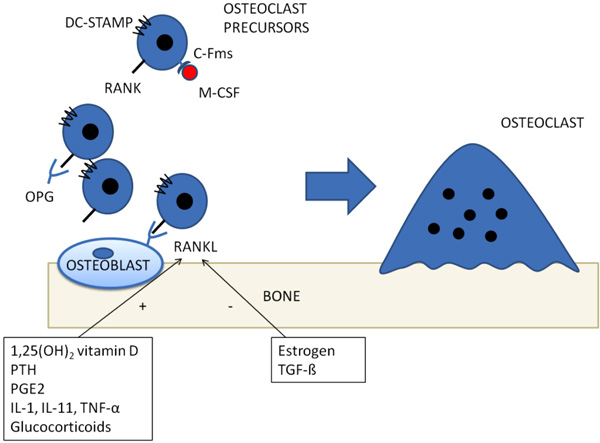
The best known molecular pathway involved in inflammatory osteoporosis is, probably, represented by Receptor Activator of Nuclear Factor Kappa B (RANK) / RANK ligand (RANKL) system. Since early ‘80s, it has been proposed that osteoblasts could regulate osteoclasts activity, based on the observation that osteoblasts, but not osteoclasts, express receptors of bone-resorbing factors, such as PTH and prostaglandin E2 [16]. In the following years, an osteoblastic transmembrane protein, called RANKL was cloned and shown to be upregulated by bone-resorbing factors [17]. It acts via binding its receptor, RANK, on osteoclasts precursor, in a cell-to-cell interaction which induces osteoclasts differentiation and activation Fig. (1) [18, 19]. A decoy receptor, which suppresses osteoclasts differentiation, has also been cloned (Osteoprotegerin - OPG) [20]. Proinflammatory cytokines, such as Tumor Necrosis Factor α (TNF-α), interleukin (IL)-1, IL-6 and IL-17, enhance RANKL mediated differentiation, proliferation and activity of osteoclasts [8, 17, 21-23]. T-cells, and Th17 in particular have the most relevant role in creating a “osteoclastogenic” environment during systemic inflammation by releasing IL-17, RANKL, TNF, IL-1, and IL-6 [8] thus enhancing osteoclasts differentiation and bone resorption.
In the last years, a direct correlation between autoimmunity and bone loss in RA has been unveiled; in fact, the classical antibodies of seropositive RA, Rheumatoid Factor and antibodies recognizing citrullinated proteins, have been proposed as potent osteoclasts inducers [24].
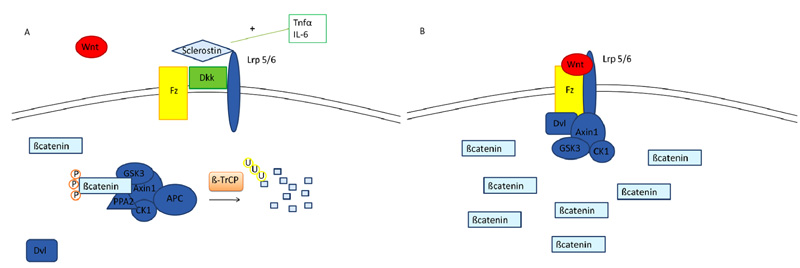
A further biological system which is involved in “inflammatory osteoporosis” is Wnt/ß catenin pathway. ß catenin is a transcriptional co-activator normally degraded in the cytoplasm by a multiprotein degradation complex, consisting of the tumor suppressors Axin and Adenomatous Polyposis Coli (APC), Ser/Thr kinases GSK-3 and CK1, Protein Phosphatase 2A (PP2A) and the E3-ubiquitin ligase β-TrCP; this complex starts the degradation process of β-catenin by targeting it for ubiquitination, after which β-catenin is subsequently digested by the proteasome [25]. The activation of the Wnt signaling pathway leads to the accumulation of ß catenin. In fact, when a member of the Wnt protein family binds the transmembrane receptor Frizzled (Fz) and its coreceptors LRP 4, 5 or 6 (lipoprotein receptor-related proteins), axin is removed from the degradation complex and β catenin is released, thus accumulating itself in the cytoplasm [26] Fig. (2). Finally, β catenin translocates to the nucleus where it links to other transcriptional factors (TCF/LEF, T-cell factor/lymphoid enhancing factor) creating a complex which enhances the transcription of target genes [27]. Several secreted protein families antagonize or modulate Wnt/β-catenin signaling: sFRPs (secreted Frizzled-related proteins) and WIF (Wnt inhibitory protein) bind to Wnt, and in the case of sFRPs, also to Fz acting as Wnt antagonists [28]. Other important Wnt inhibitors are the members of Dickkopf (Dkk) family and Sclerostin, which are LRP5 and 6 ligands/antagonists; they prevent Wnt proteins to bind to LRP5/6, thus inhibiting the Wnt/β-catenin signaling [29, 30]. Wnt signaling is essential for skeletal formation and development and is involved in chondrogenesis, in differentiation, survival and proliferation of osteoblasts [31, 32], synthesis of bone matrix as well as differentiation and function of osteoclasts during development [33]. During systemic inflammatory disease, TNF-α induces osteocytes to synthetize sclerostin and DKK-1 [34], thus negatively affecting osteoblasts activity and blocking new bone formation. In a TNF-α transgenic mouse model, DKK-1 is overexpressed, blocking osteoblasts differentiation and promotes sclerostin expression and osteocyte death [35]. Sclerostin, plays a crucial role in these activities, since its inhibition in the same model reverted the inflammation-related bone loss [36]. Likewise, IL-6 has been described to inhibit Wnt signaling in synovial fibroblasts and in osteoblasts [37].
In a study by Terpos et al. on RA patients, an osteoprotegerin/RANKL ratio 5 times lower than that observed in healthy controls has been reported, with an inverse correlation between circulating osteoprotegerin levels and disease activity (as DAS28 score) and a positive correlation between RANKL and C-reactive protein. Furthermore, DKK-1 and sclerostin levels were higher in RA patients than in healthy controls. After two months of treatment with tocilizumab (a humanized anti-interleukin-6 receptor antibody), the osteoprotegerin/RANKL ratio increased proportionally to clinical improvement and suppression of inflammation; at the same time, sclerostin concentration increased while DKK-1 decreased with respect to baseline [38]. Consistently, prospective studies have reported a protective effect of antiTNF agents on bone loss [39, 40].
The improvement of our knowledge about the mechanisms underlying bone loss, helped in the development of novel treatment strategies [41, 42]. In fact, in the last decades, new drugs were developed. RANK/RANKL system has been the first targeted; in fact, a human monoclonal antibody, denosumab, targeting RANKL is the first biological drug developed for the treatment of osteoporosis. According to the results of the FREEDOM trial, the use of denosumab twice yearly for 36 months was associated with a 68% reduction in vertebral fractures (RR 0.32, 95% CI 0.26-0.41) and a 40% reduction in hip fractures (HR 0.60, 95% CI 0.37-0.97) [43]. In the open-label extension of the FREEDOM trial, denosumab treatment for up to 10 years was associated with low rates of adverse events, low fracture incidence compared with that observed during the original trial, and continued increases in BMD [44]. Interestingly, targeting RANKL in the specific setting of RA seems to collaterally give protection against the development of erosions [45]; on this basis, denosumab might be considered a promising novel treatment for the management of RA [46].
More recently, neutralisation of sclerostin emerged as a novel therapeutic approach. Romosozumab, a monoclonal antibody anti-sclerostin, induced a significantly higher increase in BMD than alendronate, teriparatide or placebo in a phase 2 study on 419 postmenopausal women [47]. In the phase 3 trial FRAME [48], romosozumab was effective in reducing the risk of vertebral fracture after 12 months of treatment, although no effect was seen on non-vertebral fractures. Ongoing clinical trials will better clarify the clinical usefulness and safety of this molecule.
Finally, a further new drug has been recently proposed for the use in the management of osteoporosis. Odacatinib is a selective inhibitor of Cathepsin K, a protein secreted by mature osteoclasts to degrade bone matrix [49]. Although the interim results of the phase 3 study (LOFT) were promising in terms of BMD improvement [50], following the demonstration of an increased HR for strokes, Merck announced the discontinuation of the development of odanacatib [51].
3. VITAMIN D/PTH AXIS AND BONE HEALTH
Vitamin D3 is a pleiotropic hormone, the main activities of which are the result of the interaction of its active metabolite (1,25(OH)2 vitamin D3, also called calcitriol) with the Vitamin D Receptor (VDR). When 1,25(OH)2 vitamin D3 becomes available, the VDR heterodimerizes with Retinoid X Receptor (RXR) and moves from cytoplasm to nucleus, where it binds specific DNA regions (Vitamin D Responsive Elements, VDRE). VDR, indeed, acts as a transcriptional factor, either up- or downregulating many different target genes [52, 53]. Although recent evidences have related vitamin D to many different aspects of human physiology, the more relevant activity of this hormone is related to bone homeostasis and calcium/phosphate balance as a result of the actions on several targets: kidney, gut, parathyroid gland, bone.
1,25(OH)2 vitamin D3 co-participates with Parathyroid hormone (PTH) in the maintenance of calcium plasma concentration in the narrow range of normality; indeed, this has relevant implications in bone homeostasis, being bone the main storage site of calcium in human body. Calcitriol is able to increase calcium concentration, by enhancing its absorption in the gastrointestinal tract [54]; the active transcellular calcium absorption, mediated by 1,25(OH)2 vitamin D3, is a saturable process that involves the passage of calcium across the luminal brush border membrane, its transfer into the cell cytoplasm and its active extrusion through the basolateral membrane. 1,25(OH)2 vitamin D3 upregulates some key genes in calcium intake, such as calcium channel TRPV6 [55], Calbindin-D9k [56] and the basolateral active channel intestinal plasma membrane ATPase (PMCA1b) [57]. Moreover, calcitriol acts on the kidney, stimulating calcium intake by inducing the expression of key genes involved in calcium handling in tubules, like TRPV5, TRPV6 and calbindin-D28k [58].
PTH synergistically acts together with vitamin D to maintain calcium plasma concentrations. In fact, PTH secretion is regulated by Calcium Sensing Receptor (CaSR) [59]; in response to decrements in serum ionized calcium, CaSR induces the secretion of PTH which is able to directly enhance calcium reabsorption in the distal tubule through regulation of TRPV5 [60]. Moreover, it is a strong inducer of CYP27B1 the hydroxylase which mediates the last step of vitamin D activation [61].
Along with calcium plasma concentration, vitamin D also regulates phosphate metabolism. Serum phosphate levels are not as tightly regulated as serum calcium, and they rise with a high phosphorus diet, particularly after meals. Approximately 70% of dietary phosphate is absorbed in the small intestine. Phosphate absorption mainly occurs by a passive, concentration-dependent diffusion process. However, a fraction of phosphorus is absorbed through a saturable, active transport, which is facilitated by 1,25(OH)2 vitamin D3 [62]. On the contrary, PTH has a phosphaturic effect on kidney [63]. Taking together all these actions, vitamin D is responsible for an increase of calcium and phosphate plasma concentration, while PTH has hypercalcemic and hypophosphatemic effects.
More interestingly for the aim of the present review, both vitamin D and PTH directly act on bone. 1,25(OH)2 vitamin D3 acts on RANK/RANKL regulatory system. 1,25(OH)2 vitamin D3 is one of the stronger inducers of RANKL in osteoblasts [64] and, on the other hand, a suppressor of OPG synthesis [65], acting as a master regulator of physiological bone turnover. Similarly, PTH increases calcium and phosphate efflux through stimulation of RANKL by osteoblasts, which in turn stimulates osteoclast-mediated bone resorption [66]. Vitamin D and PTH interplay in a crosstalking balance. In fact, if on one hand PTH enhances vitamin D activation, calcitriol both indirectly and directly downregulates PTH production and release [67]. Moreover, 1,25 (OH)2Vitamin D3 inhibits parathyroid cells proliferation [68]. This explains why, in case of inadequate vitamin D plasma concentration, PTH plasma concentrations increase, aiming to maintain calcium concentration within physiological ranges, leading to the so-called secondary hyperparathyroidism [69].
From the previous discussion, it is evident that the maintenance of vitamin D/PTH axis is essential for bone health since both hormones have effects on the two main mechanisms leading to bone formation: bone turnover and bone mineralization, by regulating calcium/phosphate balance. Although historically well characterized, new lights have been shed on this hormonal axis after the discovery of novel mediators of bone homeostasis, which interact with vitamin D and PTH. Fibroblasts Growth Factor 23 (FGF23) was firstly identified in mouse brain in 2000 [70] and has two main physiological functions: a phosphaturic effect emanating directly from bone that coordinates bone phosphate flux and a counter-regulatory effect to vitamin D activity. It is mainly expressed by osteocytes [71] and osteoblasts [72] and its activity is mediated by FGF23 receptor together with an obligate coreceptor, αKlotho [73]. FGF23 synthesis is induced by Phosphate-regulating neutral endopeptidase X-linked (PHEX), Dentin Matrix Protein (DMP)-1, sustained phosphate load, 1,25 (OH)2 vitamin D3, low Klotho, PTH and serum ionized calcium [74]. Its main action is to counter-regulate vitamin D activity by suppressing CYP27B1 and enhancing the inactivating enzyme CYP24A1 in the kidney [75]. Moreover, FGF23 directly inhibits phosphate reabsorption in the kidney [76] and suppresses PTH synthesis by parathyroid cells [77]. Consistently with these findings, the major consequences of FGF23 excess are hypophosphatemia, aberrant vitamin D metabolism, impaired growth, and rickets/osteomalacia. Inversely, deletion of FGF23 in mice results in hyperphosphatemia, excess in calcitriol and soft tissue calcifications [78]. The discovery of FGF23 allowed us to understand that the relationship between vitamin D and bone is bidirectional; if, on one hand, vitamin D is essential for bone health, on the other hand, a bone-derived mediator is able to modulate vitamin D metabolism, generating an intricate crosstalk. The relevance of vitamin D in bone health is confirmed by clinical data; in fact, in vivo, a deficient vitamin D status and the consequent secondary hyperparathyroidism cause osteoporosis and osteomalacia, which are often undetected but lead to a decrease in BMD and to an increased risk of fractures. In young children who have little mineral in their skeleton, this defect results in a variety of skeletal deformities classically known as rickets [79]. The definition of the optimal vitamin D threshold for bone health is still largely controversial, however, a 75 nmol/l plasma level is deemed adequate by many for fracture prevention [80].
4. RA-RELATED OSTEOPOROSIS: THE ROLE OF VITAMIN D
The maintenance of a normal vitamin D status remains of paramount relevance in RA; in fact, an inadequate vitamin D status can magnify the deleterious effect of inflammation on bone density [81-83]. Moreover, some authors postulated a potential role of vitamin D as a modulator of inflammation in RA, although the evidences are still inconclusive [81]. RA patients show a decreased BMD, being therefore at higher risk for osteoporosis and fractures [84] so an impairment of vitamin D status can be claimed as a potential pathogenetic factor of RA-related osteoporosis.
First of all, a very high prevalence of hypovitaminosis D has been reported in patients affected by Autoimmune Rheumatic Diseases (ARD) in general and in RA patients specifically. Considering non-supplemented patients, the prevalence of hypovitaminosis D, in a rheumatology outpatient clinic in Northern Italy, has been reported to be as high as 87% [85]. Similar prevalences have been reported in other cohorts of rheumatic patients who were not undergoing cholecalciferol supplementation, all over the world [86-88]. However, whether ARD are independent risk factors for hypovitaminosis is highly debated; although lower plasma 25(OH) vitamin D concentrations have been described in ARD patients with respect to the general population by some authors [89-91], others did not confirm this observation [92-94]. Specifically looking at RA patients, a prevalence of 85% and 45% has been reported for insufficiency and deficiency, respectively considering the thresholds of 75 and 50 nmol/l [88, 95]. Despite these discrepancies, there is a general agreement about the association of vitamin D levels with disease activity [91, 96].
In this context it is relevant to underline the immunoregulatory actions of vitamin D. In fact, the active form of vitamin D regulates the activity of monocytes and macrophages [97], B and T cells [98-101]. Interestingly, vitamin D is able to decrease the production of crucial proinflammatory cytokines, such as IL-6 and TNFα which are involved in the pathogenesis of bone loss [102]; it could be therefore argued that the deficient vitamin D status might enhance the proinflammatory milieu characterizing RA, thus favouring the bone loss.
An indirect evidence of the implication of vitamin D in the pathogenesis of osteoporosis in RA is derived by the evidence that a VDR polymorphism has been linked to bone loss in RA [103]; in particular Rass and colleagues [104] found a lower BMD in RA patients carrying the BB and Bb genotypes of the VDR BsmI polymorphism with respect to carriers of the bb genotype. These results suggest that the B allele may be a marker for increased bone reabsorption and bone loss in RA.
Finally, a further element of interest is related to a recent observation according to which ARD patients show an impairment of vitamin D/PTH axis. In fact patients affected by inflammatory rheumatic diseases have higher plasma PTH than controls for similar vitamin D concentrations; in other words, PTH suppression seems to be more refractory to plasma vitamin D than in general population, contributing to the development of a “relative hyperparathyroidism” [105]. Consistently, PTH concentration correlates with disease activity in RA, irrespectively of vitamin D concentration; in fact, patients with erosive disease seem to show higher plasma PTH concentrations [106]. Different models might explain these findings. Chronic inflammatory processes may reduce parathyroid cells sensitivity to active vitamin D. Alternatively, immune cells might consume 1,25(OH)2 vitamin D3, at the expense of the amount available to act on bone health [105]. In fact, vitamin D can be directly activated in calcitriol by CYP27B1, which is expressed by human macrophages [107, 108] and dendritic cells [109].
The main issue is how to correct vitamin D status in RA. A guideline dealing with cholecalciferol dietary requirements and supplementation in general population has been released in 2011 by The Endocrine Society Task Force [110]. In case of inadequate vitamin D status, the Task Force suggests the use of 50,000 IU of vitamin D2 or vitamin D3 once a week for 8 weeks to achieve a blood level of 25(OH) vitamin D above 75 nmol/l, followed by a maintenance therapy of 1500– 2000 IU/d. However, this recommendation is still largely debated and not universally accepted [111]; the best regimen in the specific subset of RA patients is even less defined. It has been shown that a high loading dose of 300,000 IU, followed by a maintenance daily dose of 800–1000 IU cholecalciferol [112], could be of advantage, being more effective in inducing PTH suppression along with vitamin D normalization (Table 2). The potential advantages of this regimen need to be weighted at the light of recent findings [113] suggesting an increase in falls and fractures risk in patients treated with a high cholecalciferol dose (500,000 IU). This last observation has been recently replicated in a randomized clinical trial; although higher monthly doses of vitamin D (60,000 IU) were effective in reaching normal 25(OH) vitamin D plasma concentrations, they had no benefit on lower extremity function and were associated with increased risk of falls compared with 24,000 IU. More specific studies on RA are required to better ponder the potential risks and advantages of high doses regimens in this population [114].
| Author | Year [Cit. Number] | Main Findings |
|---|---|---|
| Zhang J, et al. | 2012 [102] | Vitamin D decreases production of IL-6 and TNFα |
| Ranganathan P, et al. | 2009 [103] | Specific Vitamin D Receptor (VDR) alleles are associated with accelerated generalized bone loss in RA |
| Rass P, et al. | 2006 [104] | The B allele of the VDR gene BsmI polymorphism is correlated with increased osteoclast and osteoblast function |
| Sainaghi PP, et al. | 2011 [105] | Presence of higher plasma PTH levels, contributing to the development of a “relative hyperparathyroidism” condition |
| Sainaghi PP, et al. | 2013 [112] | High loading dose of cholecalciferol (300.000 UI), followed by a maintenance daily dose(600-800 UI), could be more effective in inducing PTH suppression |
CONCLUSION
In conclusion, osteoporosis is the main issue in the management of RA patients, being a typical comorbidity of inflammatory arthritis. Systemic inflammation is crucial in the development of bone loss, since it causes the impairment of different biological system involved in the maintenance of bone homeostasis. Vitamin D/PTH axis is involved in bone health and different considerations support the hypothesis that vitamin D status is particularly relevant for the development of osteoporosis in RA patients. Although further studies are required to better elucidate this topic, obtaining a normal vitamin D status is paramount in preventing RA-related osteoporosis; therefore, the correction of a deficient vitamin D status should be suggested to each rheumatic patient. Currently, there is no consensus on the best regimen for these patients and further studies are required to better address this important aspect of the management of RA patients.
CONSENT FOR PUBLICATION
Not applicable
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

