All published articles of this journal are available on ScienceDirect.

Evaluation of Autoantibodies in Patients with Primary and Secondary Sjogren’s Syndrome
Authors Info & Affiliations
Abstract
Background:
Antibodies to salivary gland protein 1 (SP1), carbonic anhydrase 6 (CA6) and parotid secretory protein (PSP) were discovered in an animal model of Sjogren’s syndrome (SS). Their expression was noted in patients with SS, especially those with lower focus scores on lip biopsies and those with early disease lacking antibodies to Ro and La.
Objective:
The current studies evaluated these autoantibodies in patients with long-standing SS expressing high levels of anti-Ro antibodies and in patients with Sjogren’s syndrome secondary to systemic lupus erythematosus (SLE), systemic sclerosis (SSc) and mixed connective tissue disease (MCTD).
Method:
Sera were obtained from patients and evaluated by ELISA for IgG, IgA and IgM antibodies to SP1, CA6 and PSP.
Results:
IgA anti-CA6 antibodies were noted in 38% of these patients, but anti-SP1, CA6 and PSP IgM or IgG antibodies were identified only in a minority of patients. In patients with secondary SS, antibodies to SP1/CA6/PSP were more sensitive and specific than anti-Ro .
Conclusion:
While more studies are needed, antibodies to SP1, CA6 and PSP provide valuable markers for the diagnosis of primary and secondary SS, especially early in the course of the disease.
INTRODUCTION
Sjogren’s syndrome (SS) is a common autoimmune disease in which damage to the salivary and lacrimal glands leads to dry eyes and dry mouth and secondary problems from the lack of protective secretions. A percentage of patients with primary SS will also develop systemic manifestations including lung disease, kidney disease, peripheral neuropathy, vasculitis and lymphoma [1, 2]. SS may also occur secondary to other autoimmune diseases including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SSc), polymyositis (PM) and mixed connective tissue disease (MCTD) [3, 4].
Diagnostic criteria for SS are frequently being re-evaluated as more is learned about the pathophysiology of the disorder and markers that readily identify it. Both European and American criteria for the diagnosis of SS include anti-Ro antibodies [5]. More recently, additional antibodies recognizing specific salivary gland and lacrimal gland tissues have been identified in SS, anti-salivary gland protein 1 (SP1), anti-carbonic anhydrase 6 (CA6) and anti-parotid secretory protein (PSP) [6-8]. Studies in animal models of SS demonstrate that these autoantibodies are expressed earlier in the course of the disease than anti-Ro and anti-La [6]. Human studies completed to date have shown that these autoantibodies occur in patients with lower focus scores on minor salivary gland biopsies than patients expressing anti-Ro, in patients with SS secondary to rheumatoid arthritis and in 30-45% of patients with SS identified by clinical criteria who lack anti-Ro antibodies [7, 8]. The current studies were undertaken to evaluate anti-SP1, anti-CA6 and anti-PSP in a Belgian population of patients with primary and secondary SS. All primary SS were identified by clinical criteria and the presence of anti-Ro antibodies. Patients with secondary SS met clinical criteria with or without anti-Ro antibodies.
MATERIALS AND METHODS
Patients
Patient sera were obtained from a population of patients with autoimmune diseases followed at the department of Rheumatology in the University Hospital of Leuven, Belgium. Accepted European Criteria were used for the diagnosis of SS, SLE, SSc, PM and MCTD [9-13]. The treating physicians identified the presence of clinical symptoms and signs consistent with SS. Patients with primary SS were routinely followed up in the clinical department and questioned about the presence of sicca symptoms at every routine visit. Patients that were diagnosed with SLE, SSc, PM or MCTD were sent a questionnaire assessing sicca symptoms and medication use both at the time of serum sampling and at present. The local Ethical committee approved the clinical studies. The patients included in the studies were: 1) Sjogren’s 3 male and 34 female, ages 25-87 years (mean 63 years), 2) SLE 1 male and 16 females, ages 38 – 73 years (mean 52 years); 3) SSc 3 males and 11 females ages 45 – 77 years (mean 63 years); 4) MCTD 2 males and 9 females ages 27 – 72 years (mean 58 years). Normal controls were obtained from the Buffalo, New York (USA) area under a protocol approved by the Institutional Review Board SUNY at Buffalo School of Medicine. There were 24 females and 18 males ages 19 – 58 (mean 34 years).
Serological Testing
Autoantibodies to SP1, PSP, CA-VI, Ro and La were determined using ImmuLisa ELISAs (Immco Diagnostics Inc., Buffalo, NY). In brief, kits contain micro-wells coated with highly purified recombinant antigen (SP1 or PSP or CA-VI or Ro or La) or native antigens (RF). 96 plate wells are blocked and stabilized by the manufacturer to reduce non-specific binding. Controls, calibrators and diluted (1:100) patient serum are incubated in the antigen-coated wells to allow specific binding of autoantibodies to the antigen. Unbound serum components are washed off and bound antibodies are detected after incubations with anti-human IgG conjugated to HRP (Horse Radish Peroxidase) and specific enzyme substrate (TMB) as per manufacturer’ s protocol. The chromogenic reaction is stopped and the intensity of the color change, which is proportional to the concentration of the bound autoantibody, is read by a spectrophotometer at 450 nm. Results are expressed in ELISA units per milliliter (EU/ml) using the calibrators provided in the kit and reported as positive for a value > 20 or negative for a value < 20.
Statistical Analysis
Statistical analysis was performed using Prism version 7.00 for Macintosh (GraphPad Software, La Jolla California USA) and unpaired student’s t tests.
RESULTS
Studies evaluating anti-SP1, anti-CA6 and anti-PSP to date have documented their expression in some patients with long-standing SS with or without anti-Ro or anti-La antibodies, but more commonly in patients with early or less severe disease [6-8, 14]. In fact, the data to date suggest that these autoantibodies may disappear as the disease progresses while anti-Ro and anti-La become more prominent. The ELISA assays done in the studies evaluated only IgM and IgG autoantibodies. In the current studies we had several goals, first to evaluate anti-SP1, anti-CA6 and anti-PSP in a different population of patients with advanced SS, second to see if IgA antibodies to these autoantigens were present in long-standing / severe disease and third to see if these autoantibodies could identify patients with other autoimmune disease and secondary SS. Previous studies in patients with rheumatoid arthritis demonstrated the ability of anti-SP1 to identify patients with secondary SS with high specificity and sensitivity [7]. In the current studies we evaluated patients with SLE, SSc and MCTD with or without secondary SS.
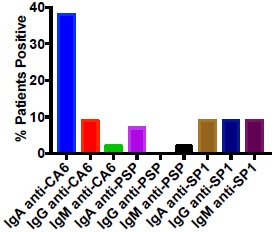
Presence of IgA Antibodies Directed Against SP1, CA6 and PSP in pSS Patients
We evaluated a population of 37 patients with long-standing SS and high titers of anti-Ro antibodies. It was anticipated that many of these patients would lack IgM or IgG anti-SP1, anti-CA6 or anti-PSP because of their long-standing disease, but we had never evaluated IgA autoantibodies in this type of patient population. Fig. (1) demonstrates as expected that few if any of the patients had IgM or IgG anti-SP1, anti-CA6 or anti-PSP. However, 38% of the patients had IgA anti-CA6 antibodies while 7% had IgA anti-PSP antibodies and 9% had IgA anti-SP1 antibodies. In a population of 42 normal controls, all patients were negative for antibodies to SP1, CA6 and PSP except one had an IgG anti-SP1, one had an IgA anti-SP1, one had an IgM anti-PSP and one had an IgM anti-CA6. The expression of IgA anti-CA6 in the SS patients was highly significant (p <0.001) compared to the normal controls, while the expression of the other autoantibodies was not significantly different.
Antibodies Directed Against CA6, PSP and SP1 in Patients with SLE, SSc and MCTD
The second study evaluated the presence of anti-CA6, anti-PSP and anti-SP1 antibodies of isotype IgG, IgA and IgM compared to IgG anti-Ro in 8 patients with SLE and no SS, 9 patients with SLE and secondary SS, 7 patients with SSc and no SS, 7 patients with SSc and secondary SS, 4 patients with MCTD and no SS and 7 patients with MCTD and secondary SS. As shown in Fig. (2), anti-Ro antibodies as well as anti-SP1, anti-CA6 and anti-PSP antibodies were seen in many patients with SLE, whether or not they had secondary SS. The differences between SLE and SLE with SS with regards to either anti-Ro or anti-SP1/anti-CA6 /anti-PSP were not statistically significant. However, a few interesting preliminary observations were made: 1) IgA anti-PSP antibodies were found only in patients with SLE and secondary SS (4 of 9); this is statistically significant compared to the normal donors (p < .001); 2) IgM antibodies to SP1, CA6 or PSP were found only in SLE patients who did not have secondary SS or developed it at the later time point; 3) of the patients with SLE but not secondary SS who had SP1/CA6/PSP autoantibodies, the most common were IgG and/or IgA anti-CA6 (5/8); and 4) of the 3 SLE patients who denied sicca syndrome at the time that the serum samples were obtained, but subsequently developed Sjogren’s syndrome at follow up (4-7 years), one had IgM anti-CA6 and anti-Ro, one had IgM anti-CA6, IgA and IgG anti-SP1 and anti-Ro and one had only IgA anti-CA6.
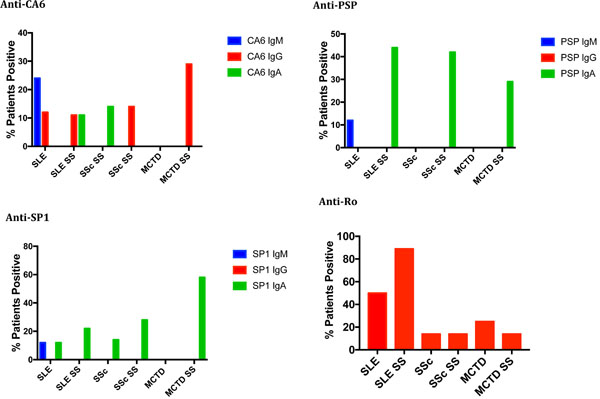
In SSc, few patients in either group expressed anti-Ro. The expression of anti-SP1, anti-CA6 and anti-PSP was noted in 71% of patients with secondary SS but only in 28% of patients without SS. The difference in expression of anti-SP1/anti-CA6/anti-PSP compared to anti-Ro in the patients with systemic sclerosis and SS was statistically significant (p = .0306) while it was not significant in the patients without secondary SS (p = .55). Interestingly, in all patients with SSc and secondary SS, the antibodies were predominantly IgA – 2 IgA anti-SP1 and 3 IgA anti-PSP; one patient had only IgG anti-CA6. Similarly, in MCTD anti-SP1, anti-CA6 and anti-PSP was identified in 58% of patients with SS but in no patients with MCTD without SS. Among the 58% expressing these autoantibodies, all expressed IgA anti-SP1, 2 also expressed IgA anti-PSP and 2 expressed also IgG anti-CA6. None expressed IgM autoantibodies. Anti-Ro antibodies were present in 14% of MCTD patients with SS and 28% of MCTD patients without SS. None of these differences were statistically significant, however, because of the small numbers of patients involved.
DISCUSSION
The current studies extend the understanding of expression of anti-SP1, anti-CA6 and anti-PSP in patients with primary and secondary SS. The first studies confirmed the fact that IgM and IgG autoantibodies to these antigens are rarely present in advanced disease. The novel finding was expression of IgA anti-CA6 antibodies in over one third of these patients. At the same time, anti-SP1, anti-CA6 and anti-PSP were shown to be potentially good markers for secondary SS in SSc and MCTD, although more patients need to be studied. Anti-Ro was less sensitive and less specific than these markers. At the same time, neither anti-Ro nor anti-SP1/CA6/PSP was good at distinguishing SLE patients with SS from those without. Although, 3 patients with SLE who initially denied sicca symptoms and subsequently developed secondary SS, all had at least one of these markers. Furthermore, certain markers, such as IgA anti-PSP were seen only in patients with SLE and SS while IgM autoantibodies were seen only in patients lacking symptoms of SS at the time the blood tests were drawn.
Evaluation of IgA autoantibodies is rarely done in autoimmune diseases, although it makes sense that disease involving mucosal areas, such as the salivary and lacrimal glands would have IgA autoantibodies. The finding of IgA anti-CA6 in advanced SS is interesting and worthy of further study. A switch from IgM autoantibodies early in the course of the disease likely resulted in IgA autoantibodies late in the course of the disease. Patients with SLE and SS, SSc and SS or MCTD with SS also had predominantly IgA autoantibodies. While larger numbers are needed to draw definitive conclusions, however, some autoantibodies, such as IgA anti-PSP were seen only in patients with an autoimmune disease and secondary SS. IgA antibodies are certainly important in various autoimmune diseases. Patients with only IgA anti-phospholipid antibodies have been shown to have the same clinical manifestations as patients with IgG or IgM anti-phospholipid antibodies [15, 16]. It is possible, however, that IgA autoantibodies may play a different role in disease pathogenesis in SS than IgM or IgG antibodies. There is some evidence in various autoimmune diseases, such as SLE that IgM autoantibodies may play a protective role and participate in tissue healing rather than playing a destructive role [17, 18]. Interestingly in our patients, IgM autoantibodies were only seen in patients who had not yet developed symptoms of dry eyes or dry mouth, although several developed them later in the course of their disease. These issues require further study.
The observation that anti-SP1/CA6/PSP identify secondary SS better than anti-Ro in SSc and MCTD is interesting and possibly related to the fact that the secondary SS developed late in the course of the underlying disease and was therefore picked up at an early stage of SS. Anti-Ro antibodies would be expected to appear later in the course of the disease. It would be interesting to evaluate the sera of these same patients at a time point two years later than the current studies to see if they developed anti-Ro antibodies. The curious observation is the frequent expression of all these antibodies in patients with SLE, whether or not secondary SS was identified. This could occur for many reasons. First, the SLE patients compared to the SSc and MCTD disease patients may have more immune activation and therefore express all autoantibodies at a higher level. Second, SLE patients tend to have more acute, severe disease manifestations than SSC and MCTD patients and may not have focused as much on dry eyes and dry mouth because they were a minor annoyance compared to their other problems. Finally, patients with SLE may all be predisposed to secondary SS but only manifest the clinical symptoms when the right combination of insults occurs. It was interesting that patients lacking sicca syndrome at the time of serum sampling but subsequently developed sicca did express one or more of the markers and often IgM autoantibodies. To put this in perspective there is an intriguing observation regarding SLE and SS made in an animal model. It was shown that the mechanisms by which the kidney is injured as compared to the salivary glands and lacrimal glands may not be the same. Eliminating marginal zone B cells from the BAFF transgenic mice getting this disorder prevents the SS but has no impact on the proliferative glomerulonephritis [19]. Perhaps a different series of events in necessary for an SLE patient to develop SS than to develop SLE alone.
CONCLUSION
In summary, these studies suggest that IgA anti-CA6 autoantibodies are frequently seen in patients with long-standing SS. They suggest that further study is needed to understand the role as well as the usefulness of antibodies to SP1, CA6 and PSP in diagnosis of secondary SS in SSc and MCTD. Previous studies suggest their role in the diagnosis of secondary SS in rheumatoid arthritis. Further studies will be necessary to confirm these observations in larger numbers of patients as well as to follow up with patients later in the course of their disease to see what has happened to the expression of these autoantibodies.
CONFLICT OF INTEREST
Long Shen and Julian Ambrus discovered anti-SP1, anti-CA6 and anti-PSP while working at SUNY at Buffalo. A patent was obtained on these autoantibodies that is held by SUNY at Buffalo and licensed to IMMCO Diagnostics, which involved with the commercial development of these assays. The SUNY at Buffalo gets royalties on this license that are shared with Julian Ambrus. Long Shen, Kishore Malyavantham and Lakshmanan Suresh all work for IMMCO Diagnostics.
ACKNOWLEDGEMENTS
Declared none.

