All published articles of this journal are available on ScienceDirect.

Felty’s Syndrome, Insights and Updates
Abstract
Felty’s syndrome (FS) is characterized by the triad of seropositive rheumatoid arthritis (RA) with destructive joint involvement, splenomegaly and neutropenia. Current data shows that 1-3 % of RA patients are complicated with FS with an estimated prevalence of 10 per 100,000 populations. The complete triad is not an absolute requirement, but persistent neutropenia with an absolute neutrophil count (ANC) generally less than 1500/mm3 is necessary for establishing the diagnosis. Felty’s syndrome may be asymptomatic but serious local or systemic infections may be the first clue to the diagnosis. FS is easily overlooked by parallel diagnoses of Sjӧgren syndrome or systemic lupus erythematosus or lymphohematopoietic malignancies. The role of genetic (HLA DR4) is more prominent in FS in comparison to classic rheumatoid arthritis. There is large body of evidence that in FS patients, both cellular and humoral immune systems participate in neutrophil activation, and apoptosis and its adherence to endothelial cells in the spleen. It has been demonstrated that proinflammatory cytokines may have inhibitory effects on bone marrow granulopoiesis. Binding of IgGs to neutrophil extracellular chromatin traps (NET) leading to neutrophil death plays a crucial role in its pathophysiology. In turn, "Netting" neutrophils may activate auto-reactive B cells leading to further antibody and immune complex formation. In this review we discuss on basic pathophysiology, epidemiology, genetics, clinical, laboratory and treatment updates of Felty’s syndrome.
INTRODUCTION
Felty’s syndrome (FS) or "Chauffard-Still-Felty Synd-rome", characterized by triad of seropositive rheumatoid arthritis (RA) with severe joint involvement, splenomegaly and neutropenia was first described in 1924 by the American physician Augustus Roi Felty [1]. FS, also referred as "extreme" or "super rheumatoid" disease, is a potentially serious systemic condition complicating long standing rheumatoid arthritis [2]. Some clinical and laboratory features of FS may have overlap with systemic lupus erythematosus (SLE), and the factors determining how life-threatening FS develops 10-15 years after a chronic course of RA still is not clear. The complete triad is not an absolute requirement, but persistent neutropenia with an absolute neutrophil count (ANC) generally less than 1500/mm3 is necessary for establishing the diagnosis [3]. FS is potentially a serious clinical condition predisposing patients to overwhelming infections and septic shock, and usually
occurs after a long course of RA although it may present simultaneously at the time of RA diagnosis [2]. Joint involvement is not necessary for diagnosis and is absent in 15-40% of FS patients. However, if it is present, joint involvement and synovial inflammation are significantly more advanced. Felty’s syndrome may be asymptomatic but clinical syndromes are usually present with local or systemic infections, and skin and pulmonary tracts are the most common sites of infection [4]. Concurrent local or systemic infections along with immunosuppression during RA management predispose patients to a potentially dangerous complication. Neutropenia is the most common and important feature of FS and splenomegaly is not always present [5, 6]. Some authors consider all RA associated neutropenias as laboratory manifestation of FS [7].
In this review we discuss on basic pathophysiology, epidemiology, genetics, clinical, laboratory and treatment updates of Felty’s syndrome.
ETIOLOGY
It is not clear that how body may lose its immune tolerance to self but the association with HLA-DR4 homozygosity and environmental factors such as immunosuppressive drugs in the context of pre-existing aggressive and seropositive RA have been described. In one prospective cohort study about 86% of FS patients were positive for HLA-DR4 [6, 8].
GENETICS
It has been shown that presence of two HLA-DRB1*04 alleles which encode shared epitopes is a predisposing factor for extra-articular manifestations of RA. There is large body of evidence that RA patients with extra-articular manifestations have worse prognosis and more mortality [9]. As mentioned before, compared to 60-70% in RA, HLA-DR4 is positive in more than 90% of FS patients. The presence of excessive HLA-DR4 homozygosity, particularly *0401/*0404 compound heterozygotes increases the susceptibility of developing FS. In a cohort of 50 patients, it has been shown that HLA-A*44, and Cw*0501 are more common in FS but HLA-B*44 is more associated with LGL [10]. One third of FS patients have clonal expansion of CD3+/ CD8+ cells in their blood denoting LGLs. These cells compose about 5% of the mononuclear cells in normal population. One-fourth of patients with these cells in peripheral blood, have arthritis with similar FS immunogenetic characteristics [11].
PATHOPHYSIOLOGY
Pathophysiology of FS associated neutropenia is multifactorial including increased neutrophil sequestration secondary to splenomegaly, peripheral destruction of neutrophils, and failure of bone marrow to produce neutrophils. It was proposed for a long time that neutropenia and splenomegaly go hand in hand in FS patients. Increased splenic sequestration and destruction of neutrophils may participate in neutropenia’s etiology, and it has been shown that splenectomy can improve neutrophil count and infectious complications [12-14]. In FS patients, spleen undergoes follicular and germinal center hyperplasia, sinusoidal plasmacytosis, increased immunoblasts and red pulp enlargement [15, 16]. Although hypersplenism has some role in the pathophysiology of FS, it does not exist in all FS patients. In a study on relation between cytokine production and clinical manifestation of FS, it has been found that serum interleukin 8 (IL8) and G-CSF were higher among other cytokines in FS patients [17]. In a study on 13 patients with FS, it has been demonstrated that FS patients with infections had higher serum level of G-CSF than patients without infections. Also this study showed that patients with FS have lower level of soluble Fcγ receptor III than patients without active infections, and may help to stratify high risk patients for infections [18]. In a case control study of 15 patients with chronic neutropenia owing to Felty’s syndrome, serum G-CSF levels were higher in FS than RA patients, 9 patients had anti G-CSF autoantibody, and presence of anti G-CSF autoantibody was higher in patients with neutropenia. This study suggested that high level of G-CSF, insensitivity of myeloid cells to cytokines, and presence of anti G-CSF autoantibody contribute in neutropenia of FS patients [19]. Interestingly, the same as FS, neutropenia is the most important and frequent clinical problem in T-LGL and is multifactorial including immune-mediated neutrophil destruction by antineutrophil antibody, and myeloid suppression by cytokines such as interferon-γ [20].
As mentioned before, large granular lymphocytes (LGLs) are activated cytotoxic T-cells (CD3+/CD8+/CD56-) and may present in a high number in peripheral blood and bone marrow of FS patients (Fig. 1 showing a LGL cell in peripheral blood).
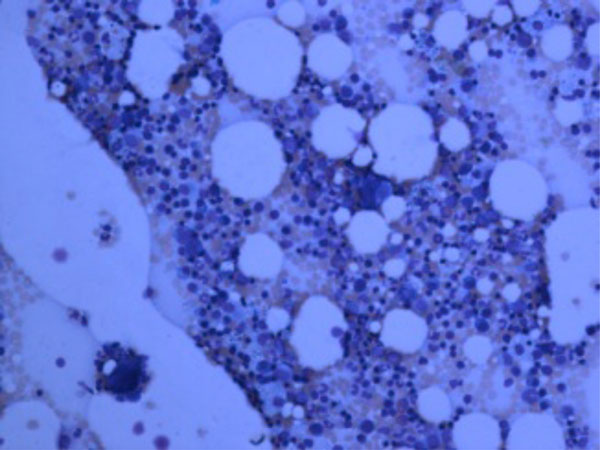
Hypocellular marrow with myeloid hypoplasia in FS.
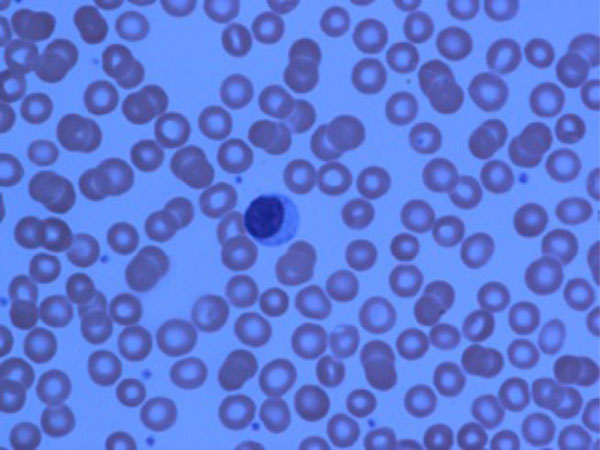
LGL cells in peripheral blood (from one of our mutual patients)
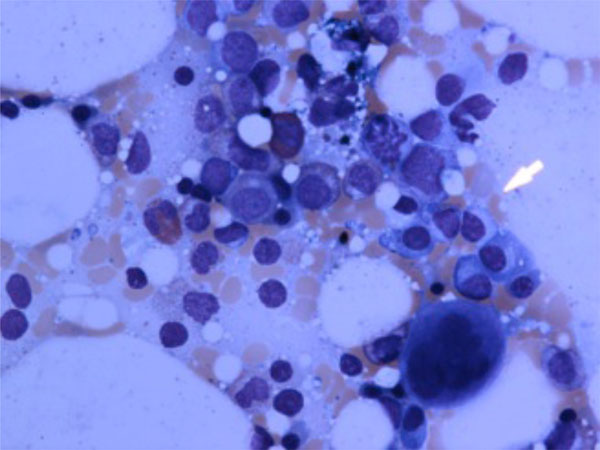
LGL cells in bone marrow.
In a cohort of 9 LGL leukemia patients with high LGL count, it has been shown that neutropenia is associated with higher grades of serum Fas ligand in 39 of 44 serum samples. This elevated serum Fas ligand was shown in rheumatoid arthritis as well. In T-LGL patients, splenomegaly and neutropenia are more common. This study suggests that higher levels of Fas ligand may increase peripheral destruction of neutrophils by inducing immune complexes and granulopoiesis suppression in bone marrow [21, 22]. Peripheral neutropenia can be associated with myeloid hyperplasia and an increase of immature myeloid cells in the bone marrow. However, myeloid hypoplasia may also be seen. Some studies have shown coating of granulocytes with immune complexes and antibodies against granulocytes and G- CSF. In a prospective study of fifteen FS patients with granulocytopenia, 11 patients had IgG antibodies against G-CSF while none of 16 patients in the control group with RA had anti G-CSF autoantibodies [20]. Binding of IgGs to neutrophil extracellular chromatin traps (NET) leading to neutrophil death plays a crucial role in pathophysiology of FS [23]. NETosis is an alternative form of cell death that is induced mostly by various types of bacterial to fungi infections [24]. NETosis causes nuclear chromatin release from neutrophils in response to diverse inflammatory stimuli including lupus autoantibodies. Neutrophil exposure to inflammatory stimuli may activate peptidylarginine deaminase 4 (PAD 4) leading to deamination of arginine-containing core histones to citrulline. There is a direct relation between peptidylarginine deaminase 4 (PAD-4) and increased susceptibility to RA. Compared to more stable PAD-4 mRNA in RA, PAD-4 in normal individuals is more neutral. As a result, RA patients may express more PAD-4. PAD-4 itself is a substrate for autoantibodies, and appearance of these autoantibodies is a sign of clinical progression of the disease. While deiminated histones are commonly seen in FS patients, they are not prevalent in RA [25, 26]. "Netting" neutrophils may activate auto-reactive B cells leading to further antibody and immune complex formation. ANCA, which has potential role in activation of neutrophils, has the properties for binding to histone which is not observed in other classic types of ANCA associated vasculitides. Chemotaxis and superoxide production is impaired. Peripheral expansion of LGL leukemia cells happens in about one-third of patients. Sinusoidal lymphocytosis, portal hypertension and nodular hyperplasia in association with hepatomegaly could be indicative of systemic vasculopathy in FS. Some studies support the same immunogenetic basis for FS and LGL leukemia and consider these two conditions as a single entity [27].
EPIDEMIOLOGY
Current data shows that 1-3 % of RA patients are complicated with FS with an estimated prevalence of 10 per 100,000 populations [28]. Comparing to the prevalence for systemic lupus erythematosus (SLE) (52 cases per 100,000 population), the relative frequency of clinical FS is actually much lower. Little information is present about the frequency of FS worldwide. It is not plausible to estimate the real incidence of FS among RA population since a large proportion of patients with FS are asymptomatic. Evidence shows that owing to wide spread use of immunosuppressive agents in RA global incidence of FS is decreasing. FS is reported to be rare in African-American patients and Caucasians are affected more than blacks [29]. Early occurrence of FS is more probable in males than females, but FS is three times more common in females than the males. HLA-DR4 genotype is strongly associated with FS, but this association is not found in LGL leukemia without arthritis. Mean age of patients with FS is within fifth to seventh decades. In a retrospective study of 165 RA patients, one patient developed FS in a five years follow up. Another retrospective study of 512 RA patients showed that 0.3% of patients had FS [30, 31].
PROGNOSIS
Owing to increased risk of serious infections in FS patients, the overall mortality is rather high and the prognosis is not favorable. This is in parallel to the increased mortality of RA in relation to extra- articular manifestations. Considering the clinical spectrum of FS from asymptomatic to a full blown devastating disease, it seems that prognostication should be made based on an individual basis. Spontaneous remission has occurred in several cases [32, 33]. In one retrospective study, although extra articular manifestations were more common in Felty’s syndrome than RA, mortality rate was similar between RA and FS [33]. However, in a prospective study of 25 FS patients, 15 patients had history of infection, and infection was the leading cause of death [34].
CLINICAL MANIFESTATIONS AND COMPLICATIONS
Although FS is a severe form of RA, it can be asymptomatic. Polyarthicular joint disease in the setting of severe seropositive RA complicated with neutropenia and infection is the hallmark of FS. Erosive RA is evident on radiographic images of involved joints. However, the involved joints are quiescent at the time of diagnosis of FS. Splenomegaly is not always present [6]. Splenomegaly can occur in RA without FS or may indicate other complications such as liver cirrhosis, brucellosis, visceral leishmaniasis, bacterial endocarditis, histoplasmosis [35] or amyloidosis. Abdominal pain can be a manifestation of splenomegaly or splenic infarcts. Weight loss may be prominent. Extra-articular manifestation of RA could be evident; hepatomegaly, lymphadenopathy, episcleritis, sicca syndrome, eye lid necrosis [36], pleuritis, neuropathy, portal hypertension, skin hyperpigmentation, leg ulcers, and vasculitis are common in FS. Liver involvement such as portal fibrosis and cirrhosis can be another extra articular manifestation of FS [37]. Infection is more common in FS than RA and skin, pulmonary tract and mouth are among the most common sites of infection. Staphylococcus aureus, gram negative enteric bacteria including Pseudomonas aeruginosa, disseminated candidiasis and other fungi are culprit organisms in FS related infections. Several mechanisms are proposed for increased risk of infection in FS. While neutropenia could be the major cause, other risk factors include high level of circulatory immune complexes [38], hypocomplementemia, leg ulcers and concomitant steroid use, impaired phagocytosis and intracellular killing, impaired chemotaxis and superoxide production may also play some role to increase the risk of infections [39, 40]. A study in 15 FS patients with absolute neutrophil count less than 2000/µl found that autoantibodies against G-CSF are associated with decreased serum Fc γ receptor III and may show increased infection index in neutropenic patients of autoimmune diseases [19]. Procalcitonin could also indicate concurrent infection in neutropenia [41]. Liver involvement is not common in Felty’s syndrome, but if present, combination of hepatic nodular regenerative hyperplasia, portal hypertension, and variceal bleeding are characteristic findings in FS [37, 42]. Overall risk of all cancers is increased in FS patients. There is a two-fold greater chance of non-Hodgkin’s lymphoma (NHL) in RA patients, which is probably directly related to the severity of RA. Patients with FS have more risk for developing NHL than RA population. This higher risk is similar to the increased risk of Sjogren’s syndrome in RA [43, 44] (Table 1).
Clinicobiologic indices in similar autoimmune disorders.
| Variable | Felty’s Syndrome | LGL Syndrome | Sjӧgren Syndrome | Systemic Lupus Erythematosus |
|---|---|---|---|---|
| Average age | 60 | 60 | 55 | 25 |
| F/M ratio | 2-3/1 | 1.5/1 | 9/1 | 9/1 |
| HLA | DR4 | Not known | Not known | DR2, DR3 |
| CD marker | Not known | 3,8,57 | Not known | Not known |
| Splenomegaly | Often | Sometimes | Rare | Not rare |
| Lymphadenopathy | Not rare | Sometimes | Rare | Not rare |
| Leukopenia | Neutropenia (Always) | Neutropenia (Often) | Rare | Lymphopenia (common) Neutropenia (sometimes) |
| Anemia | Present ACD, AIHA | Present ACD | Present ACD | Frequent ACD, AIHA |
| Autoantibody | Almost always | Sometimes | Almost always | Typical/always |
| Hypocomplementemia | Frequent | Not known | Rare | Frequent |
| Prognosis | Poor | Variable | Good | Variable |
| Secondary malignancies | Two folds | Not known | Increased | Increased |
Abbreviations: F, female; M, male; CD; Cluster of differentiation; ACD; anemia of chronic disease, AIHA; autoimmune hemolytic anemia, HLA; human leukocyte antigen.
LABORATORY INVESTIGATION
Neutropenia is the hallmark of FS. Most patients may also suffer from anemia of chronic disease and thrombocytopenia. High titers of rheumatoid factor presents in most patients. Antinuclear antibodies (ANA), anti-perinuclear antibodies, and anti-keratin antibodies can be positive in both RA and FS patients. However, these antibodies are neither sensitive nor specific to be used for diagnosis and treatment follow up. Antihistone antibodies are detectable in 83% of patients with Felty's syndrome, and presence of antihistone antibody in a known case of RA is almost always indicative of FS [23, 45, 46]. Erythrocyte sedimentation rate is constantly elevated. Complement components are usually depressed and circulatory immune complexes are usually elevated. Antineutrophil cytoplasmic antibodies (ANCA) of atypical type can be positive in 77% of patients. Recent studies have shown that immunoglobulin G autoantibodies in patients with FS, avidly binds to deiminated histones, (citrullinated histones) and neutrophil extracellular chromatin traps (NETosis) [25, 26]. These antibodies can easily differentiate subclinical FS from bland RA. Owing to strong association of antibodies against cyclic citrullinated peptide (ACPA or anti-CCP) and extra-articular manifestation of RA, high titers of ACPA is expected in FS as well. Positivity rate of ACPA in FS is not clear yet.
BONE MARROW FINDINGS
Bone marrow aspiration and biopsy are recommended to have a better assessment of the hematopoiesis and rule out bone marrow involvement by other hematological and non-hematological malignancies but bone marrow findings are not specific. Bone marrow can be normocellular but myeloid hyperplasia and maturation arrest are the most common findings. Unlike SLE, both relative and absolute granulocytopenia are common in FS. However, the granulocytopenia can be transient and spontaneous remission has been reported. In a review of 19 cases of FS, 18 out of 19 patients had myeloid hyperplasia and maturation arrest in all bone marrow aspirates. Marrow hypocellularity with reduced myelopoiesis and decreased numbers of mature myeloid cells secondary to myeloid suppression has rarely been reported (Fig. 1) [31, 33, 47].
LARGE GRANULAR LYMPHOCYTES
It is a very well-known fact that about 10-15% of normal peripheral blood mononuclear cells consists of large granular lymphocytes (Figs. 2, 3) which can be either T-cells (CD3+/CD8+/CD57+) or natural killer cells (CD3-/CD56+). Non-clonal and clonal expansion of T-LGLs can be seen in autoimmune disorders and chronic viral infections such as HIV, CMV, and EBV [48, 49]. T-LGL proliferation can be seen in RA and FS. T- LGL proliferation is a spectrum from polyclonal and reactive LGLs from one end to oligoclonal and monoclonal T-LGL lymphocytosis and overt T-LGL leukemia, which is a clonal malignant disorder with an indolent course, in the other end. TCR rearrangement can be detected by polymerase chain reaction (PCR) and Southern blot [50, 51]. It is important to emphasis that presence of oligoclonal or monoclonal proliferation of T-LGL cannot be interpreted as T-LGL leukemia since clonal expansion of T-LGLs may be a natural or pathologic immune phenomena associated with autoimmunity or viral infections [51]. The whole spectrum of LGL proliferation can be seen in FS patients, and the common presenting symptoms and signs include fatigue, fever, night sweats, weight loss, neutropenia and splenomegaly [51].
Rheumatoid arthritis is a very common finding in large granular lymphocyte leukemia associated with splenomegaly and neutropenia, and interestingly HLA-DR4 is common in both entities [52, 53]. The incidence of clonal T-LGL in RA is not well defined, and is reported in less than 1% of RA patients but this observation may be due to missed diagnosis [28]. Mutation of signal transducer and activator of transcription 3 (STAT3), an active mediator of cytokine signalling, pathway is involved in LGL leukemia, and their clonal LGL proliferation may be seen with FS patients. Demonstration of STAT3 mutation status may help to classify LGL leukemia patients and solve the diagnostic dilemma [54].
ANEMIA
Anemia in FS patients can be normocytic and normochromic secondary to the chronic disorder. Felty’s syndrome is more commonly associated with positive Coombs’ test and autoimmune haemolytic anemia than RA [55]. However, causes of anemia such as iron deficiency anemia due to chronic blood loss from drug- induced gastritis or megaloblastic anemia secondary to B12 and folate deficiency should be ruled out.
DIFFERENTIAL DIAGNOSIS
Felty’s syndrome can be a differential diagnosis of any systemic disease with neutropenia. LGL leukemia [56], Hodgkin and non-Hodgkin lymphomas are among the neoplastic differential diagnosis [16, 42]. T-LGL may behave as a chronic inflammatory disease, and is the main differential diagnosis of any RA patient who presents with neutropenia [57].
MANAGEMENT
There is no definitive treatment for FS and no randomized clinical trials available for FS. Isolated neutropenia is not essentially an indication for specific treatment unless granulocyte count < 1000/mm3. Untreated cases of RA with granulocytopenia usually respond well to conventional disease modifying anti-rheumatic drugs. Special attention should be made in caring patients with prominent constitutional symptoms (including fever) and RA. Administration of broad-spectrum antibiotics covering the most important microbial or fungal agents in conjunction with correcting underlying neutropenia is prudent. Despite traditional use and reported success of the abounded drug “gold” in correcting neutropenia and splenomegaly in FS, its use is outdated owing to extensive side effects. Low dose methotrexate (MTX), a folate antagonist inhibiting purine synthesis that is widely accepted treatment for classic RA, is the most commonly used drug as the initial treatment [58] This drug interfere with interleukin- 1 activity and leads to adenosine accumulation. Cyclophosphamide (with active metabolite phosphoramide mustard) is seldom an initial choice owing to potential role of inducing neutropenia and harboring risk of infection. While corticosteroids are empirically used in FS with good initial response, but its long term use is a major issue due to increased risk of infection and these agents should probably be viewed as a second-line treatment modality [16]. In a report of two cases of FS with severe neutropenia and methotrexate associated toxicity, hydroxychloroquine had a long lasting effect and increased the neutrophil count after a few days [59]. Leflunamide had disappointing results in FS. Continuous production of autoantibodies has been observed in FS and B cell depletion therapy with rituximab may have some potential role in FS management. Sustained remission was seen in one case report [60] but rituximab was ineffective in two men with FS in a six months follow up [61]. However, in a 46 year old woman with 13 year history of RA, and newly developed FS who failed to respond to treatment a 12 months therapy with rituximab resulted in a PMN response [62]. Other biological agents mainly TNF- alpha blocking agents such as etanercept, infliximab, and adalimumab were used in 6 patients with FS, and none of them led to sustained improvement in neutrophil count [63]. Owing to increased risk of neutropenia and infection, these biological agents are not good options.
GRANULOCYTE COLONY STIMULATING FACTOR
There is no randomized clinical trial study to treat FS neutropenia and its infection complications with colony stimulating factors. However, several case reports have been shown that granulocyte colony- stimulating factor (G-CSF) is an effective therapeutic modality in the management of severe neutropenia in FS [64-66]. There is large body of evidence that G-CSF may be associated with flu-like symptoms, exanthema, thrombocytopenia, hyperuricemia and increased alkaline phosphatase. Vasculitic skin rash and exacerbations of arthritis following G –CSF usage in FS patients have been reported. There are several case reports that G-CSF may induce autoimmune disorders flare and skin rash. In a patient with FS and profound neutropenia associated with recurrent infections, G-CSF significantly increased neutrophil count after 24 hours. However, G-CSF was stopped owing to vasculitic maculopapular rash and RA flare up. In a 57 year old male with RA, splenomegaly and pneumonia, G-CSF should be discontinued after five days owing to severe bone pain regardless of significant increase in polymorphonuclear leucocytes [67-69]. Biopsy proven leukocytoclastic vasculitis induced by G-CSF was complicated a FS patient who 11 presented with occipital scalp abscess. In this case, 15 days after starting subcutaneous G-CSF for severe neutropenia, patient developed tender purpuric lesions in his lower extremities. Patient responded well to prednisone and G-CSF stopped [70]. G-CSF was used in a 46 year old female with FS and maturation arrest of the neutrophil in bone marrow but it was stopped after patient developed joint inflammation and leukopenia. After rechallenging with G-CSF, patient experienced severe joint pain along with an increase in WBC count [71]. In a patient with history of two year of FS, neutropenia and recurrent soft tissue infections without any active synovitis, she developed fever, active arthritis and maculopapular rash the day of receiving G-CSF which did subside after G-CSF was discontinued [72]. G-CSF is an accepted therapeutic measure for the management of FS with severe neutropenia and infection. In patients who are undergoing splenectomy G-CSF can also be used in the management of preoperative period.
In a prospective study of 8 patients with FS, splenomegaly and neutropenia, subcutaneous G-CSF was given for recurrent or severe infections secondary to neutropenia or to cover a major surgery. Six patients had improvement in absolute neutrophil count without significant side effects [73-75]. Absolute neutrophil count was improved after a few days of starting G-CSF in a FS patient with recurrent severe infections with E coli refractory to antibiotics. The peripheral blood granulocytes increased from base line of 4% to 74% with significant drop in platelets without any clinical consequences [76]. In a case of rheumatoid arthritis, LGL and agranulocytosis, G-CSF has been used in management of the chronic neutropenia refractory to splenectomy. A bone marrow aspiration 10 days after starting G-CSF subcutaneously showed that maturation arrest of granulocytes improved with increment of neutrophils in peripheral blood [77]. In another case of LGL with severe sepsis and neutropenia refractory to G-CSF, cyclosporine was added to G-CSF. Patient had a good response to combination of cyclosporine and G-CSF and tolerated the treatment well [78]. In four patients with LGL and severe granulocytopenia without splenomegaly, human recombinant GM-CSF improved number of peripheral granulocytes both in vivo and in vitro [79]. It has been reported that GM-CSF was used successfully in a 60 year old FS patient with perianal infection and maturation arrest proven in bone marrow aspiration. However, patient developed pleural and 11 pericardial effusion and skeletal pain after administration of subcutaneous GM-CSF without improvement in peripheral neutrophil count [80]. In another case of severe recurrent infection secondary to LGL, with normal bone marrow cellularity and very few granulocytes who has been presented with severe resistant skin infection, subcutaneous GM-CSF was added to antibiotic regimen. Addition of GM-CSF did not have any additional clinical benefit [81]. However, a two weeks trial of subcutaneous G-CSF with dose escalation was unsuccessful in a 60 year old man with LGL and chronic agranulocytosis and recurrent infections. A trial of subcutaneous GM-CSF increased the number of granulocytes but medical team decided to discontinue the treatment owing to the side effects such as fever and severe bone pain [82]. Our experience in agreement with others shows that by modifying the dosage and frequency of G- CSF, it can be started at the lowest effective dose to reach the goal of ANC above 1,000/mm3. G-CSF injections once a week or biweekly usually keeps ANC >1000/ mm3.
SPLENECTOMY
Therapeutic modalities such as MTX and growth factors can be used for the management of severe neutropenia and splenectomy can be avoided in the majority of FS patients. It is important to monitor patient clinical condition rather than following laboratory values, considering this important fact that neutropenia does not predispose every patient to infectious complications. Thus, prophylactic splenectomy is not recommended and splenectomy is always the last therapeutic modality for FS patients who have severe neutropenia (ANC < 500/mm3) and frequent infections. Splenectomy can improve neutropenia, but it does not provide a long-lasting effect. Almost all patients show some improvement in neutrophil counts after splenectomy but neutropenia reoccurs in approximately 25% of the patients [13, 83]. In a retrospective study, a cohort of 15 patients diagnosed with T-LGL and rheumatoid arthritis with confirmed splenomegaly, elective splenectomy was done and patients were followed for a median of 719 days. Bi- or pancytopenias improved after splenectomy in most patients with a lower morbidity. This study suggest that splenectomy may be associated with a favorable outcomes in patients with LGL proliferations [84].
CONCLUSION
Felty’s syndrome is a challenging subtype of seropositive RA with longstanding, severe and erosive arthropathy. The complete triad of erosive RA, splenomegaly and neutropenia is not considered an absolute requirement for making the diagnosis and the mere presence of RA associated with persistent neutropenia with an ANC less than 2000/mm3 is satisfactory for establishing the diagnosis. Bone marrow aspiration and biopsy are recommended as part of neutropenia work up. T- LGL proliferation may be seen in FS patients. Felty’s syndrome increases the risk of life threatening bacterial infections of skin, mouth and respiratory tracts. Even moderate to severe neutropenia (ANC <1000/mm3) is not an indication for using agents such as MTX or performing splenectomy. Recurrent infections need to be treated. Therapeutic modalities such as methotrexate (MTX) and low dose G- CSF can be used in the management of FS patients with neutropenia and frequent infections. Splenectomy should be considered as the last resort in patients who do not respond to the above mentioned measures.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

