All published articles of this journal are available on ScienceDirect.

Human Endogenous Retroviruses (HERVs) and Autoimmune Rheumatic Disease: Is There a Link?
Authors Info & Affiliations
Abstract
Autoimmune rheumatic diseases, such as RA and SLE, are caused by genetic, hormonal and environmental factors. Human Endogenous Retroviruses (HERVs) may be triggers of autoimmune rheumatic disease. HERVs are fossil viruses that began to be integrated into the human genome some 30-40 million years ago and now make up 8% of the genome. Evidence suggests HERVs may cause RA and SLE, among other rheumatic diseases. The key mechanisms by which HERVS are postulated to cause disease include molecular mimicry and immune dysregulation. Identification of HERVs in RA and SLE could lead to novel treatments for these chronic conditions. This review summarises the evidence for HERVs as contributors to autoimmune rheumatic disease and the clinical implications and mechanisms of pathogenesis are discussed.
INTRODUCTION
Over the last three decades there has been an interest in human endogenous retroviruses (HERVs) as potential contributors towards autoimmune disease, made all the more important given that the incidence of autoimmunity is increasing worldwide [1]. Endogenous retroviruses (ERVs) and other retroviral elements have been found in all vertebrates investigated [2]. Much work has been done to elucidate the numerous retroviral families that exist within the human genome, of which ERVs make up 8% [3]. HERVs are believed to be pathogenic in several autoimmune diseases, but especially the rheumatic diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) [4, 5]. The growing body of evidence suggests RA and SLE are complex diseases which require genetic susceptibility, environmental factors and opportune circumstances operating together to induce disease [6]. Previous studies have explored the possibility of induction of SLE by Epstein - Barr virus (EBV) and HERVs [7-9].
The aim of this review is to summarise the existing evidence for the role of HERVs in autoimmune rheumatic disease, focussing specifically on RA and SLE. This paper describes several hypotheses on the mechanisms by which HERVs are thought to be pathogenic. This is important as the link between HERVs and rheumatic disease could lead to the discovery of novel therapeutic agents – and even cures - for these chronic diseases.
HERVS: ORIGIN AND CLASSIFICATION
Retroviruses are small viruses which replicate by reversing the normal flow of genetic information from DNA to RNA, known as reverse transcription. Exogenous retroviruses such as human immunodeficiency virus (HIV) and human T cell leukaemia virus (HTLV) can reproduce viral RNA from their pro-viral DNA, and hence retain infectivity, a feature lost by HERVs. HERVs were first integrated into the human genome 30–40 million years ago and are believed to be the key molecular link between the host genome and exogenous viral particles [10, 11]. Over millions of years, HERVs (coined “fossil viruses”) have become trapped within the human genome. They are transmitted genetically in a Mendelian fashion and are found within the DNA of all cells. HERVs have similar gene structures to exogenous retroviruses, and are composed of gag, pol, and env regions sandwiched between two long terminal repeats (LTRs) (see Fig. 1) [12].

Structure of human endogenous retroviruses (HERVs). HERV products may be generated using different open reading frames (ORFs)
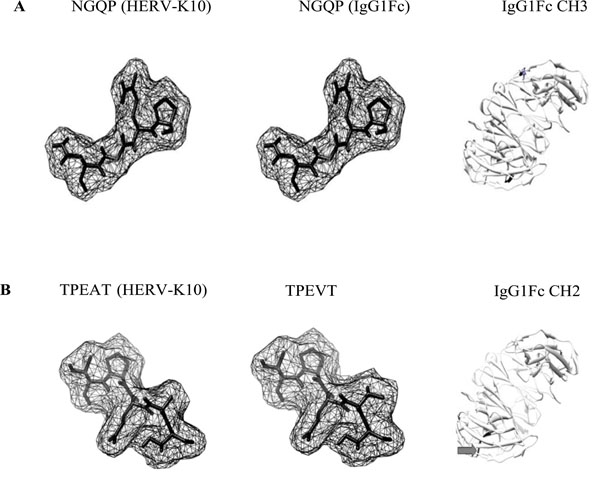
Molecular models of HERV-K10/IgG1Fc epitopes. Epitopes A and B taken from Westwood [18] and modeled using PyMOL (Molecular Graphics System, Version 1.2r3pre, Schrodinger, LLC: http://www.pymol.org/pymol) and enhanced using UCSF Chimera (http://www.cgl.ucsf.edu/chimera). Molecular models were displayed in ‘mesh’ mode. Arrows point to amino acid residues highlighted and displayed on an IgGFc molecule (PDB file: 3AVE) using UCSF Chimera. CH: constant heavy 2 and 3 domains of IgG. In B: despite the amino acid substitution, there appears to be little change in molecular shape which may facilitate antibody cross-reactivity.
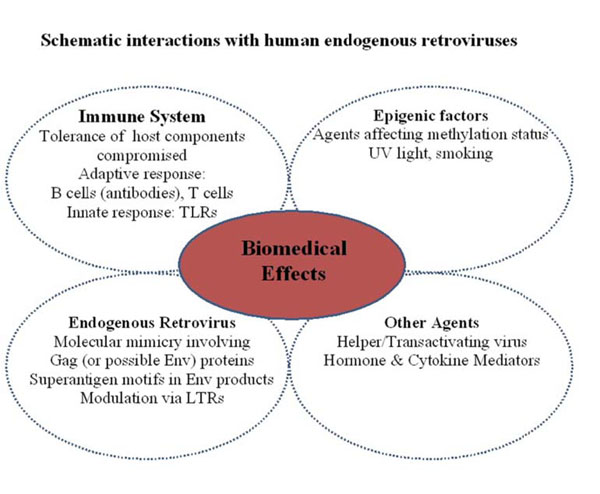
Schematic interactions with human endogenous retroviruses. The interaction of HERVs on the immune system may be complex, compounded by external and internal factors to augment detrimental effects at the molecular and protein level.
LTRs are sequences of DNA that repeat hundreds or thousands of times and are used by viruses to insert their genetic sequences into the host genomes. Gag is a polyprotein and is an acronym for Group Antigens; pol is the reverse transcriptase; and env is the envelope protein that determines viral tropism. Retroviral transcription is regulated by promoter and enhancer regions in the LTR present at both ends of the retroviral genome.
Over 26 families of HERVs have been identified [3] and, although most are defective through mutation and deletion signals, a small number have the ability to produce viral products [12]. The HERV-K family is the only family of HERVs that is able to produce intact viral particles. It is one of the most transcriptionally active families as its members retain intact open reading frames (ORFs) that encode viral particles [13]. HERVs have been broadly classified into three categories, based on their genetic similarity in the pol region (see Table 1).
| Class I | Class II | Class III |
|---|---|---|
|
|
||
| Group 1: HERV-HF | Group 1: HML-1 | HERV-L |
| HERV-H (RTVL-H, RGH) HERV-F | HERV-K (HML-1.1) | |
|
|
||
| Group 2: HERV-RW | Group 2: HML-2 | HERV-S |
| HERV-W HERV-R (ERV9) | HERV-K10 | (HERV18) |
| HERV-P (HuERS-P, HuRRS-P) | HERV-K-HTDV | |
|
|
||
| Group 3: HERV-ER1 | Group 3: HML-3 | HERV-U |
| HERV-E (4–1, ERVA, NP-2) | HERV-K (HML3.1) | |
| 51–1 HERV-R (ERV3) RRHERV-1 | ||
|
|
||
| Group 4 : HERV-T | Group 4: HML-4 | HERV-U3 |
| HERV-T (S71, CRTK1, CRTK6) | HERV-K-T47D | |
|
|
||
| Group 5: HERV-IP | Group 5: HML-5 | |
| HERV-I (RTVL-I) | HERV-K-NMWV2 | |
| HERV-IP-T47D (ERV-FTD) | ||
|
|
||
| Group 6: HERV-FRD | Group 6: HML-6 | |
| ERV-FRD | HERV-K (HML-6p) | |
|
|
||
| Other: HRES-1 | Group 7: HML-7 | |
| HERV-K-NMWV7 | ||
|
|
||
| Group 8: HML-8 | ||
| HERV-K-NMWV3 | ||
|
|
||
| Group 9: HML-9 | ||
| HERV-K-NMWV9 | ||
|
|
||
| Group 10: HML-10 | ||
| HERV-KC4 | ||
Preliminary Alignment of HERV-K10 with IgG1Fc
| HERV-K10/Autoantigen | Bioinformatic Alignment Capsid/Nucleocapsid | Viral Envelope |
|---|---|---|
|
|
||
| HERV-K10 | 370-NGQP | 3-TPEAT |
|
|
||
| IgG1Fc | 160-NGQP | 92-TPEVT |
|
|
||
| Alignment Score | 4/4 | 4/5 |
|
|
||
| Replacement Score | -12 | |
|
|
||
| Key Rheumatoid Factor Epitopes on IgG1Fc: | ||
| 256-TPEVTCVVVDVSHED-270 segment of fx2 face and b2 bend of CH2 domain | ||
| 382-ESNGQPENNYKTTPP-396 b3 bend and segment fx3 face of CH3 domain | ||
Epitopes taken from Westwood [18]. Numerals indicate amino acid position and sequence alignment (LALIGN software (http://www.ch.embnet.org/software/LALIGN_form.html)) with reference to protein accession numbers: HERV-K10 Gag2 (Capsid and Nucleocapsid) (AAA88031.1), Pol/Env (AAA88033.1), IgG Fc (AF150959). Residues highlighted in bold indicate amino acid substitutions. Alignment score shown as number of identical residues/number in peptide segment. Amino acid replacement score taken from Tudos [19]. Amino acid single letter code: G=Glycine, A=Alanine, S=Serine, T=Threonine, C=Cysteine V=Valine, D=Asparate, E=Glutamate N=Asparagine, Q=Glutamine, P=Proline, K=Lysine, H=Histidine, Y=Tyrosine. Despite a low scoring amino acid replacement for A to V, the overall molecular shape could remain similar enough to enable antibody cross-reactivity.
Summary of Studies Implicating HERVs in Autoimmune Rheumatic Disease and Molecular Techniques Used in Each
| Disease | HERV | Study | Molecular Technique | Significance/Result |
|---|---|---|---|---|
| RA | HTLV-1 p19 and HIV p24 antigens | Ziegler et al. [35, 36] | Western blot, ELISA | 45-55% patients expressed HTLV-1 antigen in synovial tissue, without serological HTLV-1 infection |
| HERV-K10 | Nelson et al. [31] | Quantitative RT-PCR | Significant association of sequence homology between HERVs and autoimmune rheumatic diseases | |
| HERV-K10 | Reynier et al. [13] | NASBA | HERV-K detected in plasma of RA patients, with higher levels observed for those with active disease. | |
| HERV-K10 | Ejtehadi et al. [46] | Multiplex RT-PCR | Enhanced expression of HERV-K10 in RA | |
| HERV-K10 HERV-W (MSRV) ERV-9 |
Nakagawa et al. [49] | RT-PCR | Multiple HERVs were expressed in normal and diseased synovium | |
| HERV-K10 | Friemanis et al. [17] | Quantitative RT-PCR, ELISA | RA patients exhibited significantly elevated levels of HERV-K gag activity compared to controls | |
| HERV-K113 | Krzysztalowska-Wawrzyniak et al. [47] | PCR | Significantly increased prevalence of HERV-K113 in patients with SLE and RA | |
| SLE | HRES-1 | Banki et al. [39] | Western blot, ELISA | Sera from patients with autoimmune disease had increased HRES-1 peptide binding activity than controls. |
| HRES-1 | Perl et al. [63] | Western blot, ELISA | Autoantibodies to HRES-1 found in autoimmune disease, especially if Ro / La negative. | |
| HRES-1 | Pullman et al. [69] | Southern blot, PCR | The HRES-1 locus at the 1q42 chromosomal region influences SLE development and disease manifestations | |
| HIAP-1 | Deas et al. [72] | Polyacrylamide gel electrophoresis (PAGE) | Cells exposed to HIAP-1 may be protected from apoptosis. | |
| HTLV-1 p19 and HIV p24 antigens | Talal et al. [37-38] | Western blot | Antibodies to p24 gag of HIV-1 found in absence of HIV-1 infection. Anti-Sm antibodies can cross-react with p24 gag. | |
| ERV-3 | Li et al. [30] | Western blot, ELISA | ERV-3 may have a role in the pathogenesis of neonatal congenital heart block. | |
| JIA | HERV-K18 | Sicat et al. [82] | Semi-quantitative RT-PCR | HERV-K18 levels elevated suggesting a mechanism for autoimmunity by superantigen stimulation of auto-reactive T cells. |
| SJS | HIAP-1 | Garry et al. [73] | PCR | HIAP-1 detected in cells exposed to salivary tissue from patients with Sjögren's syndrome. |
| HRES-1 | Brookes et al. [40] | Northern blot, ELISA | Significantly elevated levels of antibodies to HTLV-1 were found in several diseases. | |
| HERV-E env protein (λ4-1) | Hishikawa et al. [96] | Western blot | Anti-p30gag antibodies detected in sera of several diseases |
Class I contains HERVs related to Gammaretroviruses; class II viruses are related to Betaretroviruses; while Class III viruses are related to Spumaviruses [5]. Confusingly, some HERVs are known by several names, for example, HERV-K10 is also known as HML-2. A complete database of HERVs can be found online (http://herv.img.cas.cz/). It should also be noted that mobile genetic elements that behave as HERVs, such as LINE-1, have been implicated in RA [14]. LINE-1 is a DNA sequence that can change its relative position within the genome of a single cell, leading to phenotypically significant mutations. However, LINE-1 is not thought to be a true HERV by some authors as it has no LTR.
BIOINFORMATICS AND HERV RESEARCH
Bioinformatics is a rapidly-growing interdisciplinary field which harnesses computer science, mathematics, physics and biology to capture and interpret biological data [15]. Bioinformatic computer programmes can provide information about HERV protein sequences that are structurally similar (have sequence homology) to known antigenic epitopes (epitope recognition) [16]. For example, bioinformatics has been used to demonstrate that HERV-K10 shares sequence homology with epitopes of rheumatoid factor (RF) [17] (Fig. 2 and Table 2), implicating molecular mimicry (see section on “Molecular mimicry”). Short peptides that reflected these epitopes were then synthesized and an enzyme-linked immunosorbent assay (ELISA) was used to test their reactivity to patient serum. This demonstrated significant up-regulation of HERV-K mRNA levels in RA patients compared to inflammatory and healthy controls. Other molecular techniques can be used to harness HERV proteins, including: immunoblot, ELISA, nucleic acid sequence-based amplification (NASBA), polymerase chain reaction (PCR), multiplex PCR and quantitative reverse transcription–polymerase chain reaction (RT-PCR) (see Table 3). It is beyond the scope of this article to look at these in detail but it is prudent to point out that quantitative RT-PCR provides a highly sensitive technique to detect even a very low amount of RNA, found in all HERVs. Furthermore, antigen microarray profiling in RA can provide diagnostic information and allow stratification of patients with early RA into disease subsets [20].
ROLE OF HERVS IN DISEASE
Autoimmune diseases are complex diseases, whereby genetic background confers susceptibility to disease onset, but is neither sufficient nor causative for disease development [21]. The role of HERVs in disease pathogenesis is not confined to rheumatic disease, for example, it is known that expression of HERV-Fc1 is increased in patients with active multiple sclerosis [22] while in psoriatic lesions, HERV sequences of the W, K and E families are expressed and a new variant of the ERV-9/HERV-W family has been characterized [23].
The importance of genetics in the pathogenesis of autoimmune disease is supported by their clustering in families [24]. In RA and SLE a concordance rate in monozygotic twins of only 15% [25] and 25% [26] respectively is seen, emphasizing the importance of environmental factors upon the susceptibility to autoimmune disease. Genetic susceptibility might explain why only a subgroup of individuals will develop autoimmunity after infections [24]. Moreover, genome-wide analyses in lupus-prone mice have shown that major histocompatibility complex (MHC)-linked and multiple non-MHC-linked genetic factors contribute to the overall susceptibility and progression of SLE [27].
Retroviruses have been repeatedly discussed as etiological factors of autoimmune rheumatic diseases and antibodies to gag and env regions of retroviruses, including HERVs, have been reported in patients with autoimmune disease [28-31], but the role of retroviruses in these diseases remains unclear. It is known that autoimmune rheumatic disease is more prevalent in countries where retroviral infection is endemic, for example, HTLV in Japan and Central America [32, 33]. Furthermore, the observation that other types of chronic arthritis occur with an increased prevalence in HIV-1-infected individuals also lends weight to this assumption [34]. In a pivotal study of 23 patients it was demonstrated that 45% of synovial tissues obtained from patients with RA were positive for HTLV-1 p19 and p24 antigens but, intriguingly, no antibodies to HTLV-1 were detected in the sera [35]. In addition, another study showed 55% of patients with RA had p17 and p24 HIV antigens in synovial tissues but, yet again, their sera tested negative for antibodies to HIV [36]. It was therefore likely that an alternative retrovirus, with sequence homology to HTLV-1 (in the former) or HIV-1 (in the latter), was the cause of the immune response. Other studies revealed that SLE patients make antibodies to p24 gag of HIV-1 and that anti-Sm antibody can cross-react with p24 gag [37, 38]. Moreover, HRES-1 (Human T-cell lymphotropic virus-related Endogenous Sequence) is a HERV sequence capable of protein expression, and is expressed in higher quantities in patients with varying autoimmune disease than healthy controls [39, 40]. Thus, HERVs have been implicated as contributors to autoimmune rheumatic disease. Several studies have since demonstrated evidence of increased HERV presence in autoimmune rheumatic disease, summarized in Table 3.
Clinical Significance
There are potential therapeutic implications of HERVs acting as triggers for autoimmune rheumatic disease. There has been one small experimental study in 16 Sjogren’s syndrome (SJS) patients who were randomized to receive placebo or lamivudine, a reverse transcriptase inhibitor, usually harnessed as part of Highly-Active Antiretroviral Therapy (HAART) for HIV patients [41]. This experimental treatment was given on the basis of observations that the incidence of diffuse infiltrative lymphocytosis syndrome (DILS) has significantly reduced since HAART has been introduced [42]. DILS is an SJS-like illness that affects HIV-infected individuals. There was no significant difference in outcomes in this study but the small numbers may mean the study was not powered adequately. Furthermore, retroviral infections usually require combination treatment so lamivudine therapy on its own may not be effective. There are case reports of successful treatment of autoimmune rheumatic disease complications with antiviral therapy [43] but large-scale studies are needed. Of further interest is data which revealed the HRES-1 LTR is transactivated by HIV, suggesting a potential interaction between these exogenous and endogenous retroviruses. HIV stimulates expression of HRES-1/Rab4 which, in turn, abrogates recycling of CD4 to the cell surface, leading to down-regulation of CD4 expression in HIV-infected CD4+ T-cells [44, 45].
Studies have also shown higher levels of HERV-K10 expression in peripheral blood and synovial fluid in RA patients [13, 17, 46] compared with healthy controls. Higher levels of HERV expression were also observed in those with higher disease-activity scores, and hence, more active disease [13]. This may have implications for a novel biomarker in RA. Other investigators found a greater prevalence of HERV-K113 amongst RA and SLE patients; however, HERV-K113 was not associated with clinical features of disease in this study [47]. The evidence suggests that HERVs are found in greater prevalence in autoimmune rheumatic disease, which indicates a role in disease pathogenesis. Contrasting studies have shown that antibody to HERV-K10 is expressed in similar degrees in both healthy subjects and autoimmune disease [48], and that multiple HERV expression is seen in normal and diseased RA synovium [49]. Given that HERVs are incorporated into the genome, and therefore considered as self, they should not provoke an immune response. Several possible mechanisms of disease pathogenesis are described below.
HERV MECHANISMS OF AUTOIMMUNE DISEASE PATHOGENESIS
Tolerance is the process that eliminates or neutralises self-reactive immune cells [50]. Autoimmunity is caused by loss of tolerance, leading to tissue damage and destruction [51]. Tolerance is achieved in the thymus during the ontogeny of the immune system when cells that react against self are deleted (central tolerance) [52, 53]. Some potentially self-reactive cells survive these measures and are subsequently held in abeyance (peripheral tolerance) [54]. HERV-encoded proteins should be considered as self-antigens and be tolerated by the immune system but may trigger the breakdown of tolerance.
The interaction of HERVs on the immune system is likely to be complex, particularly if associated with an autoimmune disease (Fig. 3). For the endogenous virus, mechanisms such as molecular mimicry, the role of superantigen motifs and modulation via elements within LTRs have been proposed that primarily focus on branches of the adaptive immune system (B cells plus antibodies and T cells). Undoubtedly, moieties such as RNA/DNA may also contribute via toll-like receptors (TLR’s) of the innate immune system. Added to these processes, is the premise of stimulating potentially auto-reactive immune cells which have been held in abeyance through peripheral tolerance. Could this be dependent on HLA type? The breakdown of tolerance is of course a key element in autoimmunity since a number of host components may be compromised through molecular mimicry, physiochemical damage and exposure of cryptic epitopes/previously sequestered epitopes. In RA for example, IgGFc (present in all individuals) becomes a major target for autoantibodies in patients classified ‘RF-positive’, yet remains innocuous for those deemed ‘RF- negative. Other agents such as helper viruses (e.g. EBV, cytomegalovirus (CMV)) or hormones/cytokines can also facilitate the stimulation of HERVs. Presently the role of epigenetics further compounds the situation since environmental agents may surreptitiously modulate gene expression. Consequently, the interaction of external and internal factors on HERVs at the molecular level and the generation of protein products in disrupting the immune system to effect change, damage and clinical consequence will require further research.
Molecular Mimicry
The replacement of a particular amino acid may have little effect on the overall shape of an epitope. Consequently, an antibody to virus (endogenous or exogenous) could in theory cross-react with a given host protein. This scenario would be particularly important for the premise of molecular mimicry. The theory of molecular mimicry is that foreign proteins may share sequence homology with peptides on self-reactive cells, and therefore cross-activate these cells. A variety of exogenous viruses have been implicated in autoimmune rheumatic diseases, such as EBV (RA and SLE) [9, 55-57], parvovirus B19 (RA) [58] and hepatitis C (RA) [59], where molecular mimicry is central to inducing autoimmunity. Molecular mimicry is thought to be the primary mechanism by which HERVs can generate an autoimmune response and cause disease. If an HERV is present, it provides a continual source of antigen for self-reactive immune cells. A potential example of viral molecular mimicry is CMV whose genome has evolved with its host by incorporating a series of genes that functionally mimic cellular genes [60]. CMV may lead to autoimmunity through molecular mimicry, epitope spreading, and an induced immune response to cryptic antigens not normally visible to the immune system. This molecular mimicry mechanism may lead to autoimmunity in SLE patients [61, 62].
HERV antigen that is genomically similar to exogenous viral antigen may cross-react with self, and therefore be directly responsible for eliciting pathological antibodies. A key example of this is in SLE, where 70k/U1 snRNP is an autoantigen that has homology and cross-reactivity with a p30 gag protein of HRES-1 [63]. In this study, more than 50% SLE patients had anti-HRES-1 antibodies which bound to this autoantigen, compared with 3.6% healthy patients, suggesting that the autoimmune response directed against U1snRNP was triggered by the expression of HERV protein. Similarly, elevated levels of autoantibody to ERV-3 are found in pregnant patients with SJS or SLE, and transfer of maternal anti-ERV-3 antibodies across the placenta correlates with the occurrence of congenital heart block in the developing fetus [30], which further supports the pathological role of these particular antibodies. There is also evidence that HERV expression is increased by pro-inflammatory cytokines, for example, interleukin-1 up-regulates ERV-3 levels in cultured proximal tubular epithelial cells [64]. Therefore, not only could HERV infection trigger autoimmune rheumatic disease, but the resultant inflammation seen could lead to elevated HERV expression.
Altered Gene Expression and Immune Dysregulation
HERV infection could be both beneficial and detrimental to the host. Beneficial effects could include a role in blocking cellular receptors for viruses or ablating potential viral target cells, while harmful effects may be provision of a source of novel viral genes for recombination with exogenous viruses [65]. HERV sequences can integrate into any part of the genome and therefore may alter the structure and function of other genes. Retroviral integration within a gene may inactivate that gene whereas integration nearby may alter expression of the gene [66]. HERV-K10 has an integration site within the complement C2 gene, which could lead to mis-activation of the complement system [67]. Although the most pathogenic elements of HERVs are eliminated by selection, some pathogenicity may remain. HERVs could cause disease in some people and not in others due to HERV polymorphisms, as described in SLE where particular HRES-1 haplotypes exist [68]. Six inherited haplotypes of the HRES-1 locus have been defined which influence the development of SLE, one-in-particular that is a predictor for renal disease and absence of lung disease [69].
There is dysregulation of apoptosis in SLE and multiple autoantibodies exist [70] but it is unclear why so many potentially self-reactive cells survive deletion. It has been suggested that a low level of autoimmunity may be beneficial by assisting the immune system to recognize neoplastic cells by CD8+ T-cells, and thus reduce the incidence of cancer. In fact, increased survival in melanoma patients who have autoimmune disease has been demonstrated, suggesting that systemic autoimmunity may play a role in modifying the activity of established cancer [71]. Deletion of self-reactive cells via apoptosis is paramount to tolerance and evidence suggests that HERVs can modulate apoptosis. This has been demonstrated in studies involving Human Intracisternal A-type retrovirus Particles (HIAPs), which are antigenically-related to HIV and cause delayed apoptosis, and hence, prolonged foreign/self complex, enhancing the autoimmune response [72]. In a notable study, cells cultured from lip biopsies of SJS patients showed chronic infection with HIAPs [73]. Similarly, animal models have demonstrated systemic autoimmune disease in mice due to integration of an ERV within the Fas apoptosis gene, establishing a link between ERV expression, abnormal Fas expression, and autoimmune disease [74].
It is also plausible that HERVs may induce immune tolerance, as suggested by the role of syncytin in maternal immune tolerance to the fetus. Syncytin is important for placental function and has been found to derive from HERV-W [75]. It is possible that syncytin-stimulated syncytiotrophoblast formation may assure syncytiotrophoblast survival by additional interaction with immune or apoptotic mechanisms [76].
Superantigen Motifs
Superantigens are microbial proteins that are potent activators of CD4+ T-cells, which leads to excessive production of cytokines [77]. For example, bacterial superantigens causing periodontitis have been linked to development of RA [78-80]. Viral proteins may similarly act as superantigens. It is known that RA and SLE patients carry antibody to EBV in much higher frequency [9, 55-57] and EBV could act as a helper virus to HERVs. For instance, HERV-K18 possesses superantigen activity and is activated by EBV [81]. HERV-K18 levels are elevated in patients with juvenile idiopathic arthritis, suggesting a possible mechanism for autoimmunity by superantigen stimulation of self-reactive T-cells [82].
REGULATION OF HERVS VIA DNA HYPOMETHYLATION
Epigenetics refers to heritable changes in genes, regulating gene expression without changing the DNA sequence [10]. DNA methylation is an essential process for normal development and cellular differentiation. It consists of the addition of a methyl group to cytosine residues and helps to stabilize chromatin in an inactive configuration, thus inhibiting gene transcription [26, 83]. Hypomethylation can cause increased expression of given genes that are normally silenced [84]. It is thought that hypomethylation may alter synovial fibroblasts, which triggers RA development [85-86]. Altered methylation has been explored in B cells [87] and T cells [88] while changes in the pattern of DNA methylation have been found to associate with twin discordance in SLE [89]. Murine models have clearly demonstrated reduced methylation levels in the thymus of lupus-prone mice compared to non-prone mice [90]. Several other human studies have corroborated the importance of DNA hypomethylation in SLE etiology [91-93]. Expression of HERV clone 4-1 (HERV-E family) is significantly increased in SLE patients compared with healthy controls, while serum autoantibodies to this HERV and expression of its antigens on lymphocytes are detected in SLE patients, but not in healthy subjects [83, 94-96]. Furthermore, certain drugs are known to be inhibitors of DNA methylation, such as procainamide, and when demethylating agents have been given, HERV 4-1 expression is markedly increased in healthy controls, but not in SLE patients [93], which suggests involvement of demethylation of normally methylated sequences in SLE. As yet, there is no concrete link between HERVS and hypomethylation but the possibility exists. No doubt, the expanding area of epigenetics will provide the evidence required.
CONCLUSION
This paper has reviewed the evidence for HERVs as contributors to autoimmune rheumatic disease. Progress in accrual of evidence has been slow but major advances in serological, molecular and viral load have occurred. Suggested mechanisms for disease pathogenesis include molecular mimicry, immune dysregulation and superantigen motifs. The evidence appears to be mounting for HERV-K10 in RA pathogenesis and for HRES-1 and ERV-3 in SLE, but there is a clear need for animal models to corroborate this, and a call for large multicentre studies to examine the percentage of patients that exhibit high reactivity to these viruses. This may reveal other differences between populations and different races. Toll-like receptors and their possible interaction with viral material is also an emerging area of importance in both SLE and RA, which introduces the possibility that the innate system may be involved in pathogenesis. Bioinformatic and molecular modeling studies to investigate potential molecular mimicry would similarly be useful.
Given the debilitating nature and chronicity of these conditions, prevention of autoimmune rheumatic disease is the ‘holy grail’. Just as vaccination against Human Papilloma Virus is routinely given in some countries to protect females from development of cervical carcinoma, perhaps one day a similar programme may exist for those patients identified to be at high risk of developing RA (eg. individuals who are strongly RF+ and HERV-K10+). Moreover, HERV titers may one day be used as markers of disease activity or as diagnostic markers, just as RF is used today. Looking ahead, there may be a need for larger studies of antiretroviral treatment in patients with autoimmune rheumatic disease that investigate whether eliminating the viral proteins leads to disease remission. Clearly, there are numerous questions to be answered and we remain some way off this stage. National and/or international collaboration would add to the depth of quality research and help to unlock the answers within this fascinating field.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
Declared none.

