All published articles of this journal are available on ScienceDirect.

c-Jun N-Terminal Kinase in Inflammation and Rheumatic Diseases
Abstract
The c-Jun N-terminal kinases (JNKs) are members of the mitogen-activated protein kinase (MAPK) family and are activated by environmental stress. JNK is also activated by proinflammatory cytokines, such as TNF and IL-1, and Toll-like receptor ligands. This pathway, therefore, can act as a critical convergence point in immune system signaling for both adaptive and innate responses. Like other MAPKs, the JNKs are activated via the sequential activation of protein kinases that includes two dual-specificity MAP kinase kinases (MKK4 and MKK7) and multiple MAP kinase kinase kinases. MAPKs, including JNKs, can be deactivated by a specialized group of phosphatases, called MAP kinase phosphatases. JNK phosphorylates and regulates the activity of transcription factors other than c-Jun, including ATF2, Elk-1, p53 and c-Myc and non-transcription factors, such as members of the Bcl-2 family. The pathway plays a critical role in cell proliferation, apoptosis, angiogenesis and migration. In this review, an overview of the functions that are related to rheumatic diseases is presented. In addition, some diseases in which JNK participates will be highlighted.
INTRODUCTION
The c-Jun N-terminal kinases (JNKs) are members of a larger group of serine/threonine (Ser/Thr) protein kinases known as the mitogen-activated protein kinase (MAPK) family [1]. The classical JNK pathway is activated following the exposure of cells to extracellular stresses, such as UV irradiation, hyperosmolarity, and heat shock [2]. The range of initiating signals has been expanded to include a diversity of stimuli. Of particular interest is the activation of the JNK pathway following the exposure to some proinflammatory cytokines, such as tumor necrosis factor-α (TNF) and interleukin-1 (IL-1) [3]. The JNK pathway also contributes to innate immune response following ligation of various Toll-like receptors (TLR) [4]. The JNK pathway therefore acts as a critical intermediate and convergence point in immune system signaling [3].
The mammalian JNKs are encoded by three distinct genes (Jnk1, Jnk2, and Jnk3) [1]. Additional complexity is generated by alternative splicing, which results in up to 10 different protein products varying in size from 46 kDa to 56 kDa [5]. Sequence alignment of these different products shows homologies of >80%. This similarity initially suggested that the JNK proteins perform similar roles, with an equivalent ability to bind to various upstream activators and downstream substrates. However, additional complexity results from variable tissue distributions of JNK expression. JNK1 and JNK2 isoforms are ubiquitously expressed, while JNK3 is almost exclusively found in the brain, heart and testis [1]. This observation initially reinforced the idea of a specific role for JNK3 in neurons, whereas there might be redundant actions of JNK1 and JNK2 in other tissues. Yet, increasing evidence supports JNK isoform-specific roles [6].
Analogous to other MAPKs, JNKs are activated via the sequential activation of protein kinases that includes two dual-specificity MAP kinase kinases (MKK4 and MKK7) and multiple MAP kinase kinase kinases (MKKKs) such as ASK1, MEKK1-4, TAK1-3 or MLK1-3, depending on the cell lineage and the type of stimulus [2, 7] (see Fig. 1). The MKKKs phosphorylate and activate MKK4 and MKK7, which, in turn activate JNKs by dual phosphorylation. MKK4 is primarily activated by environmental stress and preferentially phosphorylates tyrosine (Y) residue 185 in the JNK1 activation motif (TPY), whereas MKK7, which is primarily activated by cytokines (TNF and IL-1), preferentially phosphorylates threonine (T) residue 183. While MKK7 is a specific activator of JNKs, MKK4 can also phosphorylate p38 MAPKs under some circumstances [8].
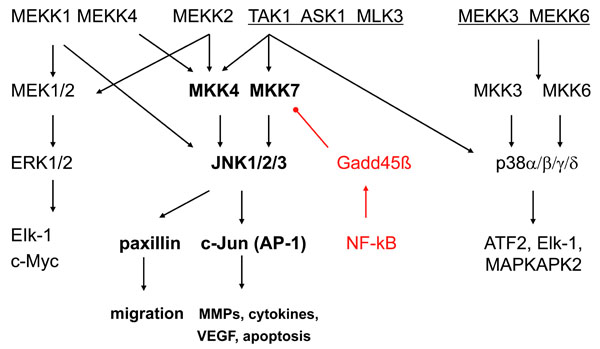
The mitogen activated protein [MAP] kinase signalling pathways. The MAPK pathways include three main families (extracellular regulating kinases, c-Jun N-terminal kinases, and p38) that form an integrating and amplifying cascade of 3 kinase tiers. JNKs are activated via the sequential activation of protein kinases that includes two dual-specificity MAP kinase kinases (MKK4 and MKK7) and multiple MAP kinase kinase kinases (MKKKs) such as ASK1, MEKK1-4, TAK1-3 or MLK1-3. The top level shows the MKKKs, the second tier shows the MKKs, and the third tier consists of the MAP kinases. Each pathway activates a variety of transcription factors that regulate genes involved with cell survival, proliferation, and inflammation. There is considerable overlap of the genes regulated by the individual MAPKs. Gadd45ß is shown as a negative regulatory mechanism that suppresses JNK by blocking MKK7. ASK, apoptosis signal regulating kinase; ATF, activating transcription factor; IL, interleukin; MAPKAP, MAPK activated protein; MEKK, MKK kinase; TAK, TGFβ associated kinase.
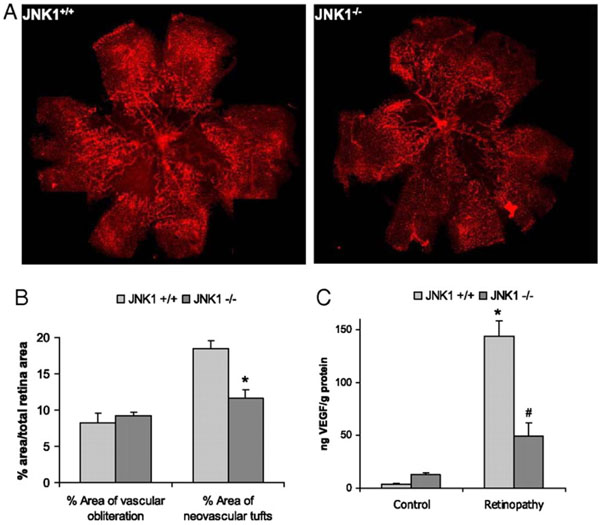
JNK1 regulates VEGF expression and neo-vascularization in a murine model of retinopathy. To determinate whether JNK1 regulates VEGF production, we analyzed its role in oxygen-induced retinopathy (OIR), a well established model of retinopathy of prematurity (ROP). In this model, when mouse pups are exposed to hyperoxia (75% oxygen) from postnatal day 7 to postnatal day 12 (from P7 to P12), vessel regression and cessation of normal radial vessel growth occur. Upon return to ambient air (normoxia) from P12 to P17, the non-perfused regions of the retina become hypoxic, resulting in expression of angiogenic factors, such as VEGF, and retinal neovascularization. Thus, litters of WT and Jnk1-/- pups were placed under hyperoxia (75% oxygen) for 5 days on P7. After 5 days, on P12, mice were returned to ambient air until P17, when they were sacrificed, their eyes were enucleated and retinas were isolated. (A) Whole mounted retinas from mice exposed to hyperoxia followed by normoxia were stained with Alexa Fluor 594-conjugated B4 isolectin from Griffonia simplicifolia, which labels endothelial cells, and viewed by fluorescent microscopy. (B) Areas of vascular obliteration and neovascular tufts were quantified using at least 6 mice per genotype. Results are expressed as means ± s.e.m. * =p< 0.05 vs WT mice (C) Retinal proteins were extracted at P17 and VEGF was quantified by ELISA. Results are averages of two experiments using at least 6 mice per genotype. Results are expressed as means ± s.e.m. * =p< 0.05 vs control; #= p< 0.05 vs WT mice [19].
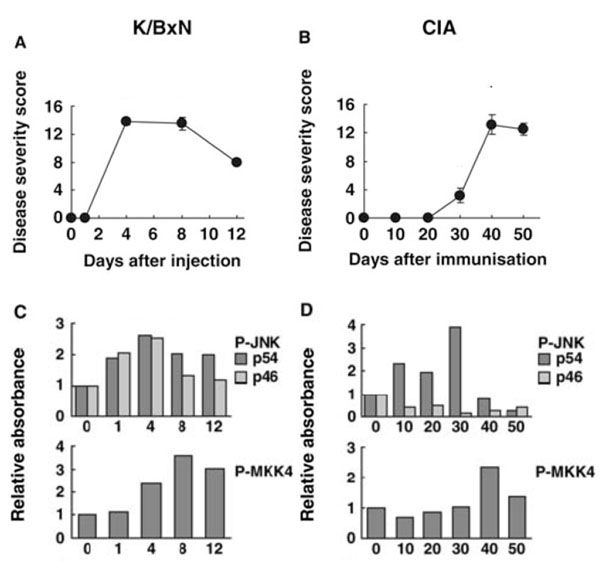
Kinetics of JNK pathway activation in joints of mice with passive K/BxN arthritis and collagen induced arthritis (CIA). The time course for JNK activation differs in two models of arthritis. Passive K/BxN arthritis is dependent only on innate immunity, while CIA involves both adaptive and innate immune mechanisms. (A) Clinical arthritis scores for passive K/BxN arthritis. (B) Clinical arthritis scores for CIA. (C and D) Phosphorylation of JNK and MKK4 were evaluated by Western blot analysis for passive K/BxN arthritis and CIA, respectively. Note that the time course for arthritis severity compared with JNK activation as well as JNK isoform activation differ in the two models [84].
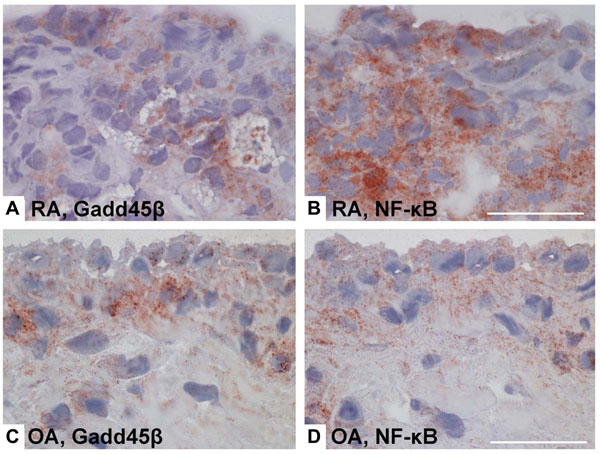
NF-κB activity and Gadd45β gene and protein expression in rheumatoid arthritis (RA) and osteoarthritis (OA) synovium. Gadd45ß is an NF-kB induced gene that suppresses the JNK pathway by binding to MKK7. Even though NF-kB activation is much higher in RA than OA, Gadd45ß expression is similar in the two diseases. Lack of Gadd45ß can contribute to over-activation of the JNK pathway in RA. (A-D), Immunohistochemical analyses of RA and OA synovium, showing Gadd45β protein expression (brown color) in RA synovial tissue (A) and OA synovial tissue (C), as well as staining for NF-κB in consecutive RA and OA sections (B and D, respectively) [86].
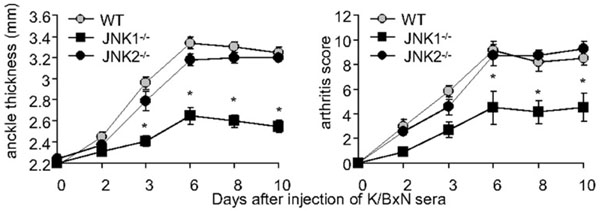
A pathogenic role for JNK1 in passive K/BxN serum transfer arthritis. Clinical arthritis scores and ankle swelling in WT (grey circles, n=15), Jnk1-/- (black squares, n=15) and Jnk2-/- mice (black circles, n=8) injected with 150 μl of K/BxN serum on day 0. Jnk1-/- mice develop less paw swelling than WT or Jnk2-/- mice. Values are means ± SEM. * =p<0.05 vs WT or Jnk2-/- mice by two way ANOVA [89].
MAPKs, including JNKs, can be deactivated by a specialized group of phosphatases, called MAP kinase phosphatases (MKPs) [2]. The MKPs are a subgroup of the protein tyrosine phosphatase (PTP) family with specificity for phosphotyrosine and phosphothreonine residues in the MAPK activation loop. Several MKPs, including: MKP-1, MKP-3, MKP-5 and MKP-7 can inactivate JNK [2]. Like other PTPs, MKPs contain a highly reactive cysteine, that mediate their enzymatic function, offering a way to regulate MAPK activity in response to production of reactive oxygen species (ROS) or exposure to thiol-reactive compounds. MicroRNA (miRNA) can also regulate MPKs. For instance, miR-101 targets MKP-1 and can regulate JNK activation in macrophages after LPS stimulation [9].
After JNKs are activated, they subsequently phosphorylate a variety of substrates that regulate a wide range of cellular functions [4]. The JNKs were originally identified by their ability to phosphorylate both Ser63 and Ser73 within the transactivation domain of the transcription factor c-Jun, which potentiates its transcriptional activity [10]. Dimerization with other Jun family members (Jun, JunB and JunD), Fos or activating transcription factor (ATF) family members comprise the transcription factor activator protein 1 (AP-1), which regulates a broad variety of genes by binding to their promoter and enhancer regions [11]. Through AP-1, JNK also regulates miRNA expression including miR-155 [12-15], known to be overexpressed in RA synoviocytes [16] and to have critical immune regulatory functions [15]. JNK phosphorylates and regulates the activity of transcription factors other than c-Jun, including ATF2, Elk-1, p53 and c-Myc and non-transcription factors, such as members of the Bcl-2 family [4]. Interactions with these nuclear and nonnuclear substrates shed light on the multiple functions of JNKs. For instance, JNKs play a role in cytokine production and extracellular matrix regulation through production of matrix metalloproteinases (MMP) [17]. JNKs functions also involve cell proliferation [11], apoptosis [18], angiogenesis [19] and migration [20]. In this review, an overview of the functions that are related to rheumatic diseases is presented. In addition, some diseases in which JNK participates will be highlighted.
JNK AND MMP REGULATION
MMP are a family of structurally and functionally related zinc-containing endopeptidases that can degrade almost all of the components of the extracellular matrix (ECM) [21, 22]. Based on their structures, sequence similarities and substrate specificities, they are often classified into several different subgroups [21, 22]. Collagenases (MMP-1 and MMP-13) that cleave the native collagen and stromelysins (MMP-3, MMP-10, MMP-11) have broad substrate specificity for proteoglycans, fibronectin and laminin and are perhaps the most important for bone/cartilage degradation. Maintaining equilibrium between deposition and degradation of the ECM is essential to normal tissue development, wound repair and tissue damage due to inflammation. In different pathological states such as inflammatory arthritis and osteoarthritis (OA), degradation is disproportionately increased with the concurrent disequilibrium [23].
MMP gene expression is primarily regulated at the transcriptional level [21, 22]. Inflammatory cytokines such as IL-1, TNF, TLR ligands, and growth factors are primary drivers of gene expression in most cell types [24]. Several transcription factors are involved in MMP gene regulation, especially activator protein-1 (AP-1) [21]. Inflammatory cytokines enhance MMP gene expression through the MAPK signaling pathway, by increasing the levels and phosphorylating different AP-1 components, such as c-Jun, JunB and c-Fos [21]. Epigenetic mechanisms, such as DNA methylation or histone acetylation, can also contribute toMMP regulation. Likewise, post-transcriptional regulatory processes including mRNA stability, protein translational efficiency, and miRNA-based mechanisms modulate MMP gene expression [21]. Recently, aberrant posttranscriptional regulation of MMPs by miRNA has emerged as an important factor in human diseases, including OA and rheumatoid arthritis (RA) [25].
The role of JNK in MMP regulation has been confirmed in cancer development, tumor cell invasion, and in fibroblast-like synoviocytes (FLS) [22]. Elevated JNK1 expression might contribute to increased MMP activity and ECM invasion by tumor cells, and also plays a major role in MMP induction in RA synoviocytes [22]. The critical role of JNK in MMP regulation in FLS was confirmed using small molecule inhibitors and JNK knockout synoviocytes in which MMP3 and MMP13 expression was significantly decreased compared to control FLS after IL-1 stimulation [17, 26]. RNA interference studies showed that only MKK7 is required for IL-1, TNF, TLR2 and TLR4 ligand-mediated JNK activation and MMP expression in cultured RA FLS, while TLR3 requires both MKK4 and MKK7 [27, 28]. After TLR3 stimulation, alternative pathways are also involved, such as IKKε, which can phosphorylate c-Jun in cultured FLS [29, 30]. The IKK-related kinase pathway could synergize with JNK by providing two parallel mechanisms for AP-1 activation. This also provides a novel link between antiviral responses, innate immunity, and destruction of the extracellular matrix in RA as TLR3 ligands have been identified in rheumatoid synovial effusions [31]. Finally, IL-1β-induced JNK activation also decreases expression of miR-27b, an miRNA that correlates with an increase of the MMP13 expression in chondrocytes [32].
Loss of articular cartilage integrity and synovial inflammation are key features of RA and OA [33, 34]. The results in genetically-modified mice, however, do not clearly support a role of MMP in extracellular matrix destruction in those diseases [35]. For instance, MMP3-deficient mice have either no difference in disease severity or paradoxical acceleration of disease in RA and OA models [36-39]. In addition, MMP inhibitors have generally been ineffective in RA and OA for clinical signs and symptoms and disease progression [40]. While an effect on MMP expression might contribute to potential benefit of a JNK inhibitor in arthritis, additional functions as described below will probably be needed.
JNK AND CYTOKINE REGULATION
Chronic inflammatory and autoimmune diseases, such as RA, psoriatic arthritis (PsA), systemic lupus erythematosus (SLE) or inflammatory bowel disease, are exemplified by imbalances in pro- and anti-inflammatory cytokines for induction of autoimmunity, inflammation and tissue damage. The cytokine milieu together with immune cell infiltration plays a key role in disease initiation and perpetuation. For example, TNF, IL-1, and IL-6 contribute to RA by increasing synovial production of chemokines, cytokines, MMPs and adhesion molecules, thereby further enhancing cell infiltration, inflammation and cartilage destruction [41, 42]. TNF, IL-1, IL-6 and IL-17 activate all three MAPK families (JNK, p38 MAP kinase, and extracellular regulating kinase (ERK)) in synovial fibroblasts [41-43], and TNF and IL-1 also activate ERK, JNK and p38 MAPK in chondrocytes [42]. The JNK pathway is also engaged in macrophages following ligation of TLRs, leading to cytokine production.
While many studies describe the activation of JNK through AP-1 in several cell types and AP-1 [44], less is known about the specific contribution of the JNK1 or JNK2 isoforms to cytokine regulation. For instance, JNK plays an important role in Tnfa gene transcription [45]. The Tnfa gene, which encodes TNF, contains JNK-responsive promoter elements required for normal Tnfa gene expression, including an AP-1 site that selectively binds heterodimeric complexes of ATF2 and c-Jun [46]. JNK can also phosphorylate ATF2 and c-Jun leading to increased transcription activity [1]. Only combined JNK1/2-deficient cells and not the single knockout cell, exhibit a severe defect in Tnfα mRNA expression [45, 47], suggesting redundant actions of JNK1 and JNK2 in TNF regulation. Although some reports suggest that JNK could also be involved in post-translational regulation of cytokines, direct mechanistic links between JNK and mRNA stability or translation remain elusive [48].
JNK AND APOPTOSIS AND CELL PROLIFERATION
JNKs are involved in cell proliferation, differentiation, survival, and programed cell death (apoptosis) [11, 49, 50]. The role of JNKs in the cellular decision to increase proliferation or cell deletion through apoptosis depends on the stimulus and the cell type [50, 51]. Sustained activation of JNK is usually associated with apoptosis, whereas the acute and transient activation of JNK may be involved in cell proliferative or survival pathway [49].
Signaling pathways that initiate apoptosis have been broadly classified into extrinsic pathways initiated by death receptors such as those of TNF, TRAIL and FAS-L, and intrinsic pathways initiated by mitochondrial events [52]. JNK has been observed to have a central role in both of these pathways. Numerous studies shows that the JNK-AP-1 pathway can stimulate expression of pro-apoptotic genes such as TNF and Fas-L. JNK also decreases the expression of pro-survival genes such as Bcl-2, Bcl-x and BAD, through multiple transcription factors in cell type- and stimuli-specific manner. The crosstalk between NF-κB and JNK pathways is also critical in TNF-induced cellular signalling pathways, which ultimately determines whether a cell lives or dies [49-51].
Apoptosis plays a critical role in many biological processes, such as embryonic development, immune responses, tissue homeostasis and normal cell turnover. For instance, apoptosis plays a central role in the immune system in the maintenance of self-tolerance and homeostatic control of lymphocyte populations [53]. Lymphocyte death is tightly controlled and there are potentially dangerous consequences when this control is abdicated, including autoimmunity. Among its many functions, JNK has a role in thymocyte negative selection, probably through c-Jun phosphorylation [3]. In the periphery, JNK might also be involved in cell death regulation during activation-induced cell death. JNK1 deficient T cells exhibit greater proliferation, which is associated with reduced activation-induced cell death [3]. This result suggests that JNK1 regulates apoptosis in T cells, as it does in other systems, although how JNK1 works has not been established.
Interestingly, JNK is not simply a proapoptotic protein kinase. It is activated by a variety of extracellular stimuli, only some of which induce apoptosis [50]. Evidence has been accumulating in certain tumors suggesting the involvement of JNK in cell survival or antiapoptosis. The molecular mechanism by which JNK suppresses programmed cell death is incompletely understood, although JNK may only exert its pro-survival function in p53-deficient cells [54].
JNK AND T HELPER CELL DIFFERENTIATION
Although only very low expression of JNK1 and JNK2 genes and their upstream kinases can be detected in naive CD4+ and CD8+ T cells [55, 56], they are induced upon activation through the TCR and, most likely, a combination of cytokines [55, 57]. After their induction in CD4+ T cells, JNK1 and JNK2 are involved in the cellular differentiation into effector T-helper 1 (Th1) and Th2 cells [3]. The expression of JNK1 and JNK2 in either Th1 or Th2 cells is similar and is much higher than naive CD4+ T cells. JNK activity can be rapidly induced in effector Th1 cells, but minimal JNK activity is detected in effector Th2 cells, suggesting that JNK might have a more important role in Th1 cells [57]. JNK1-deficient mice exhibit deficient Th1 differentiation and exaggerated Th2 responses [57]. Th2 differentiation and IL-4 production by CD4+ T cells from JNK2-deficient mice is normal, but Th1 differentiation and interferon-γ (IFN-γ) production is also impaired [58]. Other reports also suggest JNK involvement in T helper cell differentiation. MKP-7, which preferentially inactivates JNK, is induced during Th2 differentiation [59]. JNK also plays a role in IL-32-dependent maturation and activation of dendritic cells (DCs), leading to enhanced Th1 and Th17 responses as the result of increased IL-12 and IL-6 production in DCs [60].
Taken together, these data suggest that the role of JNK in T cell immune responses is to reduce proliferative responses of the activated THz cells and to potentiate polarized T cell differentiation into the Th1 and perhaps Th17 lineage [3]. JNK inhibition, therefore, could be an interesting candidate to treat Th1-mediated diseases. Although JNK1-deficient CD4 T cells have defective T cell activation or differentiation in vitro and JNK-dependent B-cell maturation and immunoglobulin production are regulated through miR-155 [13], the absence of JNK1 in T and B cells does not alter their ability to mount a pathogenic autoimmune response to myelin [61] or methylated bovine serum albumin (mBSA) [62]. The reasons for such differences are unclear, but possible explanations include the stimulation method and the complexity of an in vivo environment.
JNK AND CELL MIGRATION
Cell movement is a complex biological process requiring coordinated changes in the activities of numerous protein complexes, including actin polymerization, adhesion dynamics, and changes in cell polarity [63]. A number of focal adhesion, microtubule-associated and intermediate filament proteins (for instance, paxillin, keratin-8, tau or kinesin) are JNK substrates [4]. One of them, paxillin, was proposed as a JNK substrate based on its localization in focal adhesions and it has been studied more extensively [20]. JNK-mediated phosphorylation of paxillin is likely a key event in the regulation of migration of many cell types such as fibroblasts and cancer cells. JNK1 is also involved in macrophage migration [62].
JNK AND ANGIOGENESIS
Angiogenesis is a highly coordinated tissue remodelling process leading to blood vessel formation. Formation of new blood vessels plays an important role in the pathogenesis of rheumatic diseases, including RA and PsA, as well as malignancy and ocular angiogenic diseases [64-66]. The extent of synovial inflammation and subsequent joint destruction requires the formation of new blood vessels. Vascular endothelial growth factor (VEGF) plays a key role in angiogenesis associated with inflammation [67] and was detected in synovial fluids and tissue from RA and PsA patients, correlating with disease activity, severity and joint involvement [68, 69]. Hypoxia through the hypoxia-inducible factor (HIF) [70], and pro-inflammatory cytokines, such as TNF and IL-1 stimulate various cell types in the synovium, including macrophages, fibroblasts, vascular smooth muscle cells and synovial lining cells to release VEGF [71, 72]. Numerous other mediators including IL-6, IL-17, IL-18, nitric oxide (NO), and prostaglandins act indirectly on angiogenesis by promoting VEGF production [64].
Given the multiple roles of VEGF in regulation of vascular biology, it is not surprising that expression of this protein is tightly regulated [73]. We recently showed that another important factor involved in VEGF induction is JNK1 [19]. Mice lacking JNK1 exhibit reduced pathological angiogenesis and lower levels of retinal VEGF production in a murine model of retinopathy of the prematuity (see Fig. 2). This data indicated that JNK1 is a critical factor in hypoxia-induced retinal VEGF production and pathological ocular angiogenesis. Retinal JNK-dependent VEGF expression occured independently of HIF activation, and hypoxia stimulated JNK activity through ROS production. Once activated, JNK induced c-Jun phosphorylation at the VEGF promoter.
Other factors such as PDGF, angiopoietin/Tie2 and Fak pathways are also critically important in angiogenesis, and are highly expressed in synovial tissue [74, 75]. These pathways are involved in vessel stabilization and neoangiogenesis, and their interaction with VEGF is critical for vessel morphology and stability [76-78]. For instance, the formation of capillary-like structures during angiogenesis requires events that allow endothelial cells and pericytes to migrate into the perivascular space. FAK plays a central role in modulating endothelial and vascular smooth muscle cells migration [78]. Tie2 activation by angiopoietin promotes vessel assembly and maturation by mediating survival signals for endothelial cells and regulating the recruitment of mural cells [79]. JNK has been implicated in these pathways, suggesting that its role of JNK in angiogenesis goes beyond VEGF regulation [80, 81].
JNK AND RHEUMATOID ARTHRITIS
RA is a chronic autoimmune disease in which the importance of pro-inflammatory cytokines has been confirmed by the success of biologics that block TNF, IL-1 or IL-6. However, there remains a clinical need for new therapies, as individual regimens are only effective in a proportion of patients. Moreover, their high cost and need for parenteral administration have led to alternative strategies such as orally active small molecule. This approach could target key intracellular signaling molecules that regulate both cytokine production and cytokine action, affecting more than one cytokine or cytokine receptor.
JNK activation in RA
JNKs and its upstream kinases (MKK4 and MKK7) are highly activated in isolated RA FLS and in the rheumatoid synovial lining layer and synovial mononuclear cell infiltrates [17, 43, 82, 83]. Western blot analysis in cultured synoviocytes reveals 46 and 54 kDa species. JNK2 appears to be the dominant isoform in FLS, accounting for about 90% of the total JNK protein. Downstream, components to the AP-1 complex, c-Jun and c-Fos, are also expressed both in the synovial lining and sublining layer.
The JNK pathway in animal models
The activation and function of JNK has been extensively characterized in several animal models of arthritis, including passive K/BxN arthritis and collagen-induced arthritis (CIA) [84]. In the passive model, synovial JNK activation occurs within 1 day after injection with arthritogenic serum and before clinical arthritis occurs. Both 46 and 54 kDa isoforms are phosphorylated, peak before maximum clinical arthritis, and decline toward baseline by day 12. The kinetics of JNK activation parallels MMP3 gene expression in the joint. P-MKK4 and –c-Jun are also detected and actually reach their highest level later in the model (day 8). The results for CIA are strikingly different. Only the 54 kDa isoform is phosphorylated (unlike RA and passive K/BxN arthritis), which begins by day 10 after immunization and peaks on day 30 when MMP3 expression is also highest. Thereafter, P-JNK levels rapidly decline to baseline levels (see Fig. 3). The timing and isoform selectivity of each model needs to be considered when evaluating the potential therapeutic agents that target JNK.
Several reports suggest a critical role of JNK in inflammatory arthritis. Interference with AP-1 decoy oligonucleotides or using c-Fos/AP-1 inhibitors decreases severity of CIA accompanied by suppressing inflammatory cytokine and MMP synthesis, thus confirming the critical role of AP-1, and presumably JNK, in joint inflammation [85]. A JNK1/2 inhibitor, SP600125, was mildly anti-inflammatory in the rat AIA but it conferred striking protection against bone and cartilage destruction, together with a decrease of the collagenase (MMP-13) gene, confirming the role of JNK in arthritis [17]. In another report, deficient growth arrest DNA damage-ß (Gadd45ß) expression contributed to activation of JNK1/2, exacerbated clinical arthritis, and augmented joint destruction in passive K/BxN arthritis [86]. Gadd45ß, which is an NF-kB-regulated gene, was recently identified as an endogenous negative regulator of the JNK pathway because it blocks the upstream kinase MKK7 [87]. Gadd45ß deficiency permitted unregulated JNK activation by eliminating this endogenous negative feedback loop. Of particular relevance to RA, Gadd45ß expression is similar in rheumatoid synovium and OA tissue despite markedly higher NF-kB activation in the former [86] (see Fig. 4). Gadd45ß induction in cultured synoviocytes was also surprisingly low even when cells were activated with cytokines. These data suggest that high JNK activation in RA might be due, in part, to defective Gadd45ß expression and unregulated MKK7 function.
The role of JNK in single JNK1- or JNK2-deficient mice varies from model to model. JNK1 was found not to be essential for TNF-mediated joint disease [88]. Human TNF transgenic (hTNFtg) mice, which develop inflammatory arthritis, were intercrossed with JNK1-deficient mice. Histological and clinical analyses revealed no differences in the quantity of synovial inflammation and bone erosions or in the cellular composition of the synovial infiltrate. Moreover, cartilage damage, as indicated by proteoglycan loss in the articular cartilage, was comparable in the two strains. In other report, JNK2-deficient mice exhibited a modest decrease in bone and cartilage damage in passive collagen-induced arthritis but no difference in clinical benefit as determined by arthritis scores [26]. Levels of AP-1 activation and MMP-13 gene expression were not decreased in JNK2-deficient mice, suggesting that additional signal transduction pathways, such as JNK1, might also regulate MMP in the joint, and could induce MMP-13 expression in the absence of JNK2. Some of these data suggest that both isoforms should be targeted for effective therapy.
We recently observed new roles of JNK1 in arthritis [62, 89]. JNK1 and not JNK2 was critical for joint swelling and destruction in both the serum transfer model of arthritis (passive KxB/N) (see Fig. 5) and the antigen induced model of arthritis (AIA). The arthritogenic function of JNK1 was exerted in bone marrow derived cells, particularly mast cells in the passive KxB/N model and macrophages in the AIA model. Without JNK1, mast cells failed to degranulate efficiently in some circumstances, and released much less IL-1. We also showed that without JNK1, the ability of macrophages to migrate was impaired, even in the presence of potent chemokine stimulation. Pharmacologic JNK inhibition effectively prevented arthritis onset and abrogated joint swelling in established disease in both models. Hence, JNK1 controls certain types of mast cell degranulation, and macrophage migration and might be therefore an attractive therapeutic target in inflammatory disorders.
JNK and apoptosis in RA
In RA, the synovial environment promotes survival of FLS and discourages their deletion through apoptosis [90]. NF-κB, which is highly activated in RA and lining cells, provides a strong pro-survival signal linking inflammation and decreased apoptosis [91]. The p53 tumor suppressor, which can be mutated by the genotoxic synovial environment [92, 93], does not efficiently direct damaged synoviocytes to apoptosis. As mentioned, JNK may exert its anti-apoptotic function in p53-deficient cells. Fas-dependent synoviocyte cell death is also inefficient, but it is dependent on JNK and AP-1 when it does occur [94].
JNK and angiogenesis in RA
The observation that JNK plays a role in angiogenesis and especially VEGF expression implicates this kinase in maintaining the vasculature in inflamed synovium. Strategies to inhibit VEGF activity such as soluble VEGF-R1 or VEGF blocking antibodies reduce disease severity in murine CIA and the transgenic K/BxN mouse model of arthritis [95, 96]. In humans, these compounds have been administered to cancer patients, and VEGF or VEGFR inhibition has also been introduced to the treatment of neovascular eye diseases and recently also to arthritis trials [97, 98]. Inhibition of angiogenesis with a JNK inhibitor as a adjuvant therapy might therefore be of interest as treatment option for RA.
JNK and cell migration in RA
Migration of leukocytes into the synovium, which is partially regulated by the JNK pathway, is a multistep process involving interactions between leukocytes and endothelial cells and cellular adhesion molecules, as well as between leukocytes and chemokines and chemokine receptors. Synovial tissue and synovial fluid from RA patients contain increased concentrations of several chemokines such as monocyte chemoattractant protein-4/CCL13, monokine induced by interferon-γ/CXCL9, stromal cell-derived factor 1/CXCL12, monocyte chemotactic protein 1/CCL2, macrophage inflammatory protein 1α/CCL3, and fractalkine/CXC3CL1 [99, 100]. Targeting individual chemokines through their receptors, such as CCR1 or CCR2, has met with limited success thus far [101, 102]. Given the role of JNK in chemokine signalling, chemotaxis, and cell migration, a strategy that targets the converging intrinsic mechanisms of macrophage migration in the MAPK pathway might be effective in inflammatory arthritis.
Upstream JNK kinases in RA
To identify potentially more inflammation-specific upstream targets in the JNK MAPK pathways, synovial fibroblasts were examined for expression and activation of MAPKKs and MAPKKKs that regulate JNK MAPK signaling was studied in FLS. Although MKK4 and MMK7 are activated by IL-1, only MKK7 is essential for IL-1-stimulated JNK activation and consequent c-Jun phosphorylation and AP-1 activation in synoviocytes. These data, especially in combination with the Gadd45ß studies, suggest that targeting of MKK7 could allow selective suppression of harmful pathogenic inflammation whilst leaving MKK4-mediated activation of stress-related pathways intact [87].
Even further more upstream, the MAPKKKs most relevant to JNK activation in synoviocytes have been identified [93]. Western blot analysis and qPCR demonstrated that TAK1, MEKK1, and MEKK2 are the most abundant MAP3Ks in cultured FLS. Surprisingly, the relatively JNK-specific MEKK4 was either not expressed or was present in very low levels. Using either small interfering RNA knockdown or knockout FLS, TAK1 deficiency significantly decreased P-JNK, P-MKK4 and P-MKK7 induction but did not affect p38 activation. These results showed that TAK1 is a critical pathway for IL-1β-induced activation of JNK and JNK-regulated gene expression in FLS. In contrast to other cell lineages, MEKK1, MEKK2, and MEKK3 did not contribute to JNK phosphorylation in FLS after IL-1 stimulation.
JNK AND PSORIATIC ARTHRITIS
There is a very little information on the activation of JNK and its upstream kinases in PsA. JNK is phosphorylated in the synovium of patients with PsA and is observed mainly in the intimal lining. Some sublining mononuclear cells and the perivascular compartment also contain P-JNK [103].
Interestingly, an epidermis-specific, double knockout mouse lacking JunB and c-Jun, but not the single knockout, develops skin alterations that resemble lesions observed in patients with psoriasis [104]. As mentioned above, JNKs phosphorylate c-Jun very efficiently, although they do not phosphorylate JunB. The double-mutant mice showed a strong phenotype with inflamed plaques affecting primarily the ears, paws and tail, with a thickened epidermis, hyperkeratosis, and parakeratosis and increased subepidermal vascularization. Intraepidermal T cells, epidermal microabscesses and the typical inflammatory cell infiltrate consisting of neutrophils were seen together with increased numbers of macrophages in the dermis. Strikingly, arthritis strongly reminiscent of PsA was also observed with 100% penetrance. Inflammatory infiltrates were present in the joint regions along with massive bone destruction and periostitis. Different manifestations of the disease, such as synovitis, dactylitis and enthesitis were also recognized. Increased expression of TNF and TNF-dependent cytokines contributed to disease development, as in the absence of TNFR1 the skin phenotype was improved and arthritis was prevented.
JNK AND SYSTEMIC LUPUS ERYTHEMATOSUS
SLE is a systemic autoimmune disease associated with aberrant activation of T and B lymphocytes. Abnormal activation of intracellular signalling molecules in lymphocytes by inflammatory cytokines has been proposed as a trigger of the SLE inflammation. Several studies show high levels of JNK activation in peripheral lymphocytes in SLE patients [105]. The expression of active JNK in peripheral blood mononuclear cells significantly correlated with SLE disease activity index [106]. The data in SLE patients are also supported by data in murine models. In the parent-into-F(1) mouse model of lupus-like chronic graft-versus-host disease (GVHD), increased JNK activation was observed in splenocyte from chronic GVHD mice [107]. Another study demonstrated over-expression of P-JNK in splenocytes from (NZBxNZW)F1 female mice with established lupus in comparison to disease-free mice, which correlated with a significantly higher T cell apoptosis [108]. However, additional data are needed to understand the function of high JNK kinase activity in SLE.
JNK IN OSTEOARTHRITIS
OA is characterized by degeneration of articular cartilage secondary to an imbalance between the synthesis and integration of ECM proteins and the degradation of both synthesized ECM proteins [109]. Growth factors exemplified by insulin-like growth factor-1, its binding proteins and TGF-βcontribute to anabolic pathways including compensatory biosynthesis of extracellular matrix proteins. Catabolic pathways are altered by cytokine genes such as IL-1 and TNF, which are expressed in OA synovium. In addition, IL-1 and TNF suppress ECM protein biosynthesis while concomitantly increasing MMP gene expression [35, 110]. Chondrocytes plays a central role in maintaining the cartilage homeostasis. The vitality of articular cartilage is also critical and it can be judged on the basis of the capacity of chondrocytes to resist apoptosis [111]. Thus, OA therapeutic strategies designed to modulate the imbalance between anabolic and catabolic pathways in OA might include neutralizing cytokine activity or MMP gene expression, or inhibiting signaling pathways that result in apoptosis [109].
JNK inhibition could be a potential therapeutic candidate in OA as it plays a key role in cytokine production, MMP gene regulation and apoptosis. Surprisingly, there are little or no data of the role of JNK in animal models of OA. JNKs and the key upstream activators of JNK, MKK4 and MKK7, are expressed and activated in OA synovial tissues although their level activation is much less than in RA [17, 83]. JNKs are also expressed and activated in OA chondrocytes [112-114]. Cytokines, such as IL-1 and TNF, but also compression and mechanical stimulation engages the JNK pathway in these cells.
Although JNKs regulate MMP expression in OA chondroctyes, proteases in the ADAMTS family appear to be more relevant to cartilage degradation in OA [115]. In a murine model of OA, there was a significant reduction in the severity of cartilage destruction in the ADAMTS5-deficient mice compared with wild type mice, suggesting that ADAMTS5 is the primary aggrecanase responsible for aggrecan degradation in OA [116]. Of interest, JNK also participates in ADAMTS regulation [117]. Further studies are needed to determine if JNK in chondrocytes regulates ADAMTS expression.
Apoptosis is also a possible pathogenic mechanism in OA [111, 118]. Osteoarthritis cartilage degeneration is largely a process of destruction and failure of the ECM. Anabolic activity, phenotypic stability, and finally, survival of the chondrocytes are essential for the maintenance of proper articular cartilage. Lacunar emptying is a typical feature of osteoarthritic cartilage and has led to the assumption that cell death is a central feature in cartilage degeneration. TNF, IL-1 and NO might contribute to induction of chondrocyte apoptosis [111, 118]. Several reports suggest that inhibiting JNK signaling in chondrocytes inhibits apoptosis induced by NO and could help preserve the cartilage extracellular matrix in OA [109, 119].
CONCLUSIONS
Despite the success of biological therapies in inflammatory arthritis, orally active small-molecule drugs are alternatives for patients with arthritis for whom conventional treatments have failed or who are unable to take biological agents. They might also be effective in other diseases such as OA, where the development of effective disease-modifying drugs is urgently needed. Small molecule inhibitors have been the focus of intense efforts, with some recent notable successes [120, 121] but also some failures such as selective p38α inhibitors that have limited efficacy [122]. Selective JNK1 inhibitor could reduce destructive inflammation in autoimmune disease, especially ones mediated by Th1 cells or mast cells. However, the general trend for greater and greater specificity might be counterproductive owing to the redundancy of signaling networks. As many JNK functions are redundant between JNK1 and JNK2, the inhibition of both JNK isoforms would likely have pluripotent effects, not only on hematopoietic cells but also on FLS, endothelial cells or chondrocytes, hence being more effective. The safety of signal transduction inhibitors needs to be carefully evaluated in non-oncologic indications. However, experience with a variety of compounds in rheumatoid arthritis, including those that target Syk, JAK, p38, MEK1/2, or c-Kit, suggest that the toxicity can be managed. Thus, inhibition of JNKs or less selective approaches that inhibit more than one kinase (e.g., combination JNK–p38 inhibitor) are reasonable possibilities for novel therapeutic agents.
ACKNOWLEDGEMENTS
Supported in part by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR47825 GSF) and the Arthritis Foundation (MG)
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.

