All published articles of this journal are available on ScienceDirect.

The p38 MAPK Pathway in Rheumatoid Arthritis: A Sideways Look
Abstract
The p38 mitogen-activated protein kinase (MAPK) signaling pathway has been strongly implicated in many of the processes that underlie the pathology of rheumatoid arthritis (RA). For many years it has been considered a promising target for development of new anti-inflammatory drugs with which to treat RA and other chronic immune-mediated inflammatory diseases. However, several recent clinical trials have concluded in a disappointing manner. Why is this so, if p38 MAPK clearly contributes to the excessive production of inflammatory mediators, the destruction of bone and cartilage? We argue that, to explain the apparent failure of p38 inhibitors in the rheumatology clinic, we need to understand better the complexities of the p38 pathway and its many levels of communication with other cellular signaling pathways. In this review we look at the p38 MAPK pathway from a slightly different perspective, emphasising its role in post-transcriptional rather than transcriptional control of gene expression, and its contribution to the off-phase rather than the on-phase of the inflammatory response.
ACTIVATION OF P38 MAPK
In mammals four members of the p38 MAPK family are encoded by discrete genes. MAPKs p38α and p38β are more than 70% identical to one another, widely expressed and have essential functions in the regulation of inflammatory responses (for excellent recent reviews of the p38 pathway see [1-3]. The δ and γ isoforms are more distantly related (60% identical to p38α ), restricted in expression and are thought to have specialised functions in muscle or nerve cells. However, they may be expressed in the inflamed joint, as discussed below. Here we refer to MAPK p38α unless otherwise stated. Like the related extracellular signal regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) pathways, the p38 MAPK pathway is constructed around a three-tiered phosphorylation cascade. Activation of each MAPK requires phosphorylation at both threonine and tyrosine residues of a T-X-Y motif within a flexible domain known as the activation loop. This induces a structural alteration that catalytically activates the MAPK and increases its affinity for substates. The X residue that separates the threonine (T) and tyrosine (Y) phospho-acceptor sites is a defining feature of each MAPK family, and is glycine in the case of all the p38 MAPKs. In most cases MAPK kinase (MKK) 3 or 6 is responsible for the activating phosphorylation of p38 MAPKs. In turn, these MKKs are activated via phosphorylation by MAPK kinase kinases (MAPKKKs or MAP3Ks). Several MAP3Ks are capable of activating the p38 MAPK pathway, but in the context of immune and inflammatory responses the most important appears to be TAK1 (transforming growth factor β-activated kinase). Protein ubiquitination is now known to play a major role in the transmission of activating signals from cell surface receptors to TAK1 [4-8].
Ubiquitin is a highly conserved, abundant peptide of 76 amino acids, which can be covalently linked to proteins, in most cases at lysine residues. Ubiquitin itself contains seven lysines (at positions 6, 11, 27, 29, 33, 48 and 63), and can serve as a substrate for addition of further ubiquitin molecules, allowing the construction of long poly-ubiquitin chains. Chains that are built by sequential linkage of ubiquitin molecules through lysine 48 (K48-linked chains) usually signal for protein degradation by the proteasome. In contrast, K63-linked polyubiquitination is often a means of promoting interactions between proteins and propagating signals. Ubiquitination is carried out by an enzymatic cascade having three components known as E1, E2 and E3. An E1 enzyme activates ubiquitin and transfers it to an E2 (ubiquitin conjugating) enzyme, which then passes it to an E3 enzyme (ubiquitin ligase), which in turn mediates substrate-specific ubiquitination. In mammals there are two or more E1, approximately 50 E2 and approximately 600 E3 enzymes. There are also approximately 100 deubiquitinating enzymes (DUBs), possessing different substrate specificities and mechanisms of regulation [9]. A number of these have the property of inhibiting signaling pathways by reversing the formation of K63-linked polyubiquitin chains.
Initiation of p38 MAPK signaling is largely a matter of recruiting and activating ubiquitin ligases, and different cell surface receptors have evolved slightly different means of accomplishing this. Fig. (1) illustrates some features of cell signaling downstream of the 55 kD TNF receptor (p55 or TNFR1). For greater detail and for discussion of the mechanisms employed by different receptors to initiate p38 MAPK signaling, the interested reader is directed towards excellent recent reviews [4-8, 10]. The key step is the generation of K63-linked polyubiquitin chains, which serve as scaffolds for the oligomerization of TAK1. Such oligomerization is dependent upon the TAK1 binding proteins (TABs), which possess polyubiquitin binding domains [11]. TAK1 proteins that are brought together at polyubiquitin chains are hypothesized to phosphorylate and activate one another. They are then competent for activation of MKKs 3, 6 and other kinases. An emerging concept is that efficient activation of MAPK cascades may require a proteolytic step to release TAK1 from multiprotein signaling complexes at the cell surface. This was demonstrated for the receptor CD40, a member of the TNF receptor superfamily [12], but may also be true of other receptors that mediate activation of p38 MAPK. The coupling of MAPK activation and release of TAK1 from the receptor complex is perhaps a mechanism for limiting the duration of the response.
p38 MAPK can also be activated by self phosphorylation at threonine 180 and tyrosine 182. This non-canonical mechanism is conceptually challenging, because it requires a complete change in function from a proline-directed serine or threonine kinase to a dual specificity threonine and tyrosine kinase that has no requirement for an adjacent proline [13]. Autocatalytic activation of p38 MAPK is promoted by its interaction with TAB1, or by the phosphorylation of tyrosine 323 in response to activation of the T cell receptor [14-16]. It is likely that either TAB1 binding or tyrosine 323 phosphorylation induces a structural alteration that alters the catalytic specificity of p38 MAPK and overcomes steric barriers to self phosphorylation. In principle p38 MAPK may serve different functions when activated non-canonically, because the interaction with TAB1 or the phosphorylation of tyrosine 323 may also affect its subcellular localisation or ability to recognize substrates [17]. Another important point is that non-canonical activation of p38 MAPK may not be sensitive to the action of DUBs. However, the role of non-canonical activation of p38 MAPK in arthritis is unclear.
Once activated, either by canonical or non-canonical means, p38 MAPK proceeds to phosphorylate its own substrates in order to bring about changes in expression of pro-inflammatory and other genes. Many of the effects of the p38 MAPK pathway on gene expression are indirect, mediated by phosphorylation and activation of downstream kinases. MAPK-activated kinases MK2 and MK3 are uniquely activated by p38 MAPK, whereas the mitogen- and stress-activated kinases MSK1 and MSK2 can be activated by either p38 MAPK or ERK. Roles of these kinases in the control of inflammatory responses are discussed below.
REGULATION OF PRO-INFLAMMATORY GENE EXPRESSION BY P38 MAPK
Several transcription factors can be directly phosphorylated and activated by p38 MAPK to bring about changes in expression of pro-inflammatory and other genes. As reviewed in detail elsewhere, these include activating transcription factor 2 (ATF2, which forms heterodimers with members of the activating protein 1 [AP-1] family), signal transducer and activator of transcription 1 (STAT1, which mediates transcriptional regulation by interferons), members of the ternary complex family, which control expression of c-fos, an AP-1 family member. A necessary note of caution is that sites of phosphorylation may be targeted by kinases other than p38 MAPK, and the physiological relevance of p38-mediated phosphorylation is unclear in some cases.
The link between the p38 MAPK pathway and the function of the transcription factor nuclear factor êB (NF-κB) has been controversial for more than a decade. Inhibitors of p38 MAPK did not block the NF-κB signaling pathway but impaired the activation of NF-κB-dependent genes and reporter constructs. The MSKs, which can be activated by p38 MAPK, offer two potential solutions of this paradox [18, 19]. At least at some NF-κB-regulated genes, the phosphorylation of the NF-κB subunit p65 at serine 276 is required for productive transcription [20-23]. The MSKs are amongst the kinases that can perform this phosphorylation, providing a potential means for the p38 MAPK pathway to influence transcription of NF-κB-dependent genes. The variety of kinases that can phosphorylate p65 S276 means that p38-mediated transcriptional regulation via this mechanism is likely to be highly gene-specific and context-dependent. The p38 MAPK pathway was also found to promote recruitment of NF-κB to certain pro-inflammatory promoters, which was accompanied by phosphorylation of histone H3 at serine 10 [24]. The MSKs are amongst several kinases that can phosphorylate H3 S10. In any case, promoter-specific recruitment of MSKs may enhance transcriptional activation by NF-κB either by modifying chromatin structure to increase NF-κB recruitment, or by post-translational modification of an NF-κB component. MSK recruitment to pro-inflammatory promoters has been demonstrated [25, 26], but it is so far unclear how this recruitment occurs, or what is the causal link between the two MSK-mediated phosphorylations. The MSKs are also known to phosphorylate and activate members of the ATF/CREB (cAMP response element binding protein) family of transcription factors [18, 19].
Many reviews of p38 MAPK function emphasize these transcriptional effects, but post-transcriptional effects are at least as important in the regulation of inflammatory responses. Genome-wide analyses reveal that only half of the changes in gene expression during a stereotypical immune response can be accounted for by transcriptional regulation [27-29]. The remainder are dependent on changes in mRNA stability. The modulation of mRNA stability is a powerful mechanism for bringing about rapid changes of gene expression, such as those that occur when the innate immune system first encounters a pathogen [30, 31]. As we will describe below, defects in post-transcriptional control can contribute to inflammatory pathologies. First, we will give a brief outline of post-transcriptional regulatory mechanisms and how they are influenced by the p38 MAPK pathway.
The information content of an mRNA resides within the open reading frame, whilst the non-coding regions (or untranslated regions, UTRs) at the 5’ and 3’ ends of the mRNA convey instructions for its localization, translation and degradation (Fig. 2). Determinants of mRNA stability are most often located within the 3’ UTR, the best characterized being adenosine/uridine-rich elements (AREs). AREs contain repeats of the sequence AUUUA, and are recognized by mRNA destabilizing proteins including tristetraprolin (TTP) and several others. TTP recruits a large complex of deadenylases and associated proteins to remove the poly(A) tails of mRNAs that it is bound to [32-35]. In most cases the removal of the poly(A) tail is rapidly followed by degradation of the mRNA body.
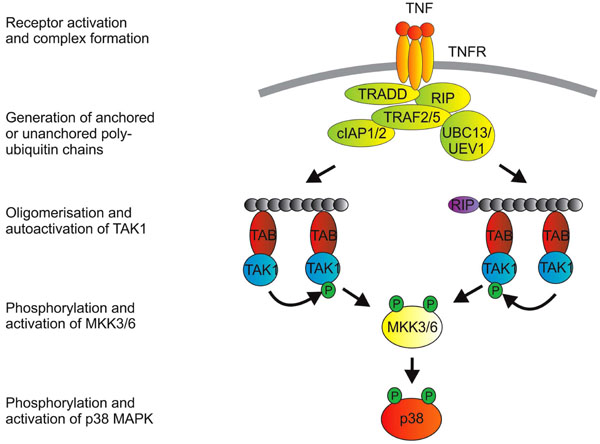
Activation of p38 MAPK downstrem of TNFR1. Engagement of TNFR1 by trimeric TNF promotes a structural change in the cytoplasmic domain of the receptor (which is itself trimeric). The cytoplasmic death domain of TNFR1 then mediates recruitment of the adaptor protein TRADD (TNF receptor associated death domain protein). This provides a platform for recruitment of additional proteins that include the kinase RIP1 (receptor interacting protein 1), TNF receptor associated factors (TRAFs) 2 and 5, cellular inhibitors of apoptosis proteins (cIAPs) 1 and 2 and, it is thought, the E2 enzymes UBC13 and UEV1. From here, the picture of signaling events becomes a little blurred, because: i) a number of entities in the complex possess ubiquitin E3 ligase activity (namely the TRAFs and the cIAPs); ii) some ubiquitin modifications of proteins in the complex have not been consistently identified in different labs; and iii) downstream events have often been studied in the context of signaling to NF-κB or JNK rather than p38 MAPK. K63-linked polyubiquitination of the TRAFs themselves or of RIP have been implicated in downstream signaling. A more recent notion is that transmission of the signal depends on the generation of unanchored polyubiquitin chains [166]. TAK1 is next recruited to the complex via interactions between polyubiquitin chains and TAK1 binding proteins (TABs), which possess poly-ubiquitin binding domains [11]. TAK1 proteins that are brought together at polyubiquitin chains then phosphorylate and activate one another. K63-linked ubiquitination of TAK1 itself may also be involved in the activation of p38 MAPK [167].
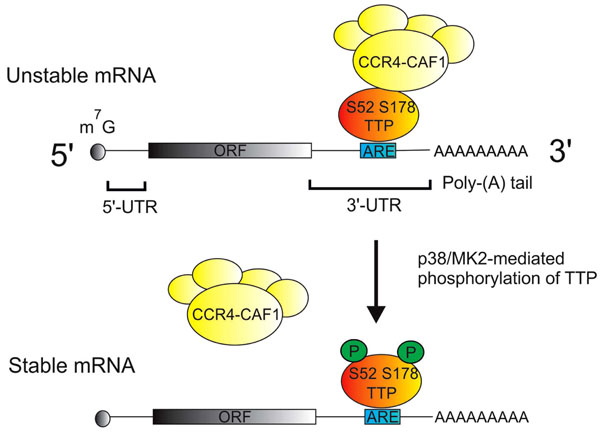
Regulation of mRNA stability by p38 MAPK and TTP. Most mRNAs are modified by the addition of a 7-methyl guanisine (m7G cap) at the 5' end, and a poly adenosine or poly(A) tail at the 3' end. The protein coding open reading frame (ORF) is flanked by 5' and 3' untranslated regions (UTRs). Adenosine/uridine rich elements (AREs) in the 3' UTRs of many cytokine mRNAs are specifically recognized by tristetraprolin (TTP), which then recruits the CCR4/NOT deadenylase complex. This complex catalyzes the removal of the poly(A) tail and initiates the degradation of the mRNA body. Following its activation by p38 MAPK, MAPK-activated protein kinase 2 (MK2) phosphorylates serines 52 and 178 of TTP, preventing the recruitment of the deadenylase complex and thus stabilizing mRNAs that are recognized by TTP.
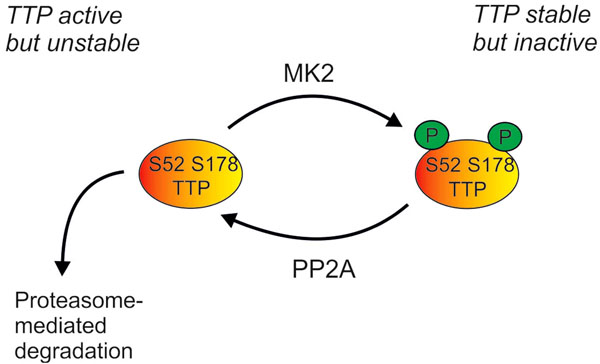
Regulation of cellular levels of TTP protein by the p38 MAPK pathway. Phosphorylation of TTP by MK2 renders it inactive (as described in Fig 2), but also protects it against proteasome-mediated degradation. TTP can be dephosphorylated by protein phosphatase 2A (PP2A), and perhaps by other phosphatases. Cellular levels of TTP are therefore dependent on a dynamic equilibrium of protein synthesis, proteasome-mediated degradation, MK2-mediated phosphorylation and PP2A-mediated dephosphorylation.
The earliest experiments with p38 inhibitors implicated a role for the p38 MAPK pathway in the regulation of translation of pro-inflammatory mRNAs, which is still emphasized in many review articles. However, an overwhelming majority of more recent papers instead point to a role for p38 in the regulation of mRNA stability (see references in [36, 37]). Genetic and biochemical experiments in a number of labs have elucidated how TTP-mediated mRNA turnover is influenced by the p38 MAPK pathway [38, 39] (Fig. 2). When activated by p38 MAPK, the downstream kinase MK2 phosphorylates murine TTP at serines 52 and 178 (S60 and S186 of the human protein) [40, 41]. This phosphorylation impairs the ability of TTP to recruit the deadenylase complex, and therefore stabilizes mRNAs that are targeted by TTP [33]. The kinase MK3 is closely related to MK2 and appears to serve a similar function in mRNA stabilisation, although it may be less strongly expressed than MK2 [38, 42]. Many mRNAs that are relevant to inflammatory arthritis are destabilized by p38 MAPK inhibitors (for example TNF, Cyclooxygenase-2, IL-1b, IL-6) [36, 37, 43-47]. In Ttp-/- mouse macrophages several of these pro-inflammatory mRNAs are constitutively stable and insensitive to p38 MAPK inhibition [48]. Therefore the inactivation of TTP is the major mechanism of mRNA stabilization by the p38 MAPK pathway in these cells. Other mechanisms of regulation of mRNA stability by p38 MAPK have also been described, and may be more significant in cell types other than macrophages [30, 37].
The likely relevance of this post-transcriptional mechanism to rheumatoid arthritis has been well illustrated by a number of genetically-modified mouse models. Mice expressing a wild-type human TNF transgene were healthy, whereas those expressing a mutated human TNF transgene with a heterologous, non-ARE 3’ UTR developed polyarthritis [49, 50]. This finding pointed to a role for the 3’ UTR in restricting TNF expression. A more elegant experiment by the same group involved the germline deletion of the ARE from the mouse TNF locus [51]. The resulting ΔARE mouse again overexpressed TNF and developed erosive arthritis (amongst other inflammatory and autoimmune pathologies). In the ΔARE mouse, TNF expression was insensitive to p38 inhibition. The TTP knockout mouse also has a complex inflammatory syndrome that includes erosive arthritis and features of autoimmunity [52]. This syndrome is largely due to an increase in the stability of TNF mRNA and the expression of TNF protein in macrophages [53, 54]. Other pro-inflammatory genes are also disregulated in the absence of TTP protein, and may contribute to the phenotype [55].
INACTIVATION OF P38 MAPK
The timely inactivation of the p38 MAPK pathway is necessary to prevent excessive inflammatory responses. The most efficient mechanism of p38 MAPK inactivation is dephosphorylation, in particular by dual specificity phosphatases (DUSPs) that catalyze removal of both threonine and tyrosine phosphates [56, 57]. The DUSP family contains approximately ten members, several of which are able to dephosphorylate and inactivate p38 MAPK, at least in vitro or under conditions of overexpression. DUSP1 appears to have a particularly important role as a regulator of inflammatory responses [58-60]. Mouse Dusp1-/- macrophages display exaggerated or prolonged activation of p38 MAPK and JNK in response to pro-inflammatory stimuli, and overexpress a variety of pro-inflammatory factors. Dusp1 knockout mice are healthy and fertile under sterile conditions, but catastrophically over-respond to pro-inflammatory insults, for example injection of endotoxin, induction of an allergic response or infection with gram negative or positive bacteria. Most relevant to this discussion, they develop a more aggressive form of collagen-induced arthritis than wild type control mice [61] (our unpublished observations).
Mathematical models suggest that modulating the activity or expression of phosphatases is a powerful means of controlling the strength and duration of activation of MAPK pathways, and hence the cellular response in terms of gene expression [62]. Consistent with its role as a central regulator of inflammation, DUSP1 expression is influenced by a wide variety of pro- and anti-inflammatory factors [58-60]. In the majority of resting cells DUSP1 is very weakly expressed, but is strongly and transiently upregulated by many pro-inflammatory agonists, its expression temporally coinciding with the off-phase of p38 activity. IL-10 may enhance or prolong DUSP1 expression in order to impair p38 MAPK signaling [63], whilst interferon γ may inhibit DUSP1 expression to enhance p38 MAPK signaling [64].
For approximately sixty years, glucocorticoids (GCs) have been used to treat rheumatoid arthritis and other chronic immune-mediated inflammatory diseases [65-67]. They possess genuine disease-modifying activity, but their use is limited by unpredictable and sometimes severe side effects [68, 69]. The upregulation of DUSP1 and the consequent inhibition of p38 MAPK is emerging as one mechanism that underlies anti-inflammatory effects [70-72]. GCs have been shown to impair p38 MAPK function in many cell types, but in Dusp1-/- macrophages they neither inhibit p38 MAPK nor strongly inhibit expression of several pro-inflammatory mediators, including TNF [73]. In experimental models of sepsis or localized acute inflammation, therapeutic effects of GCs were dependent on DUSP1 [73, 74]. Our preliminary data suggest that anti-inflammatory effects of GCs are also mediated by DUSP1 in collagen-induced arthritis. The interesting question is whether GCs depend upon this anti-inflammatory mechanism in man. Small clinical studies have suggested that the GC-induced upregulation of DUSP1 and inhibition of p38 MAPK are both impaired in severe and/or GC resistant asthma [75-78]. No equivalent study has been published in rheumatoid arthritis.
Macrophage migration inhibitory factor (MIF) is a cytokine thought to contribute to several inflammatory pathologies [79-81]. It is upregulated by GCs but antagonizes their anti-inflammatory effects, leading to the suggestion that MIF neutralization might enhance GC efficacy. Pro-inflammatory effects of MIF are thought to be mediated by inhibition of expression of DUSP1 and consequent enhancement of p38 MAPK signaling [82-85].
THE P38 MAPK PATHWAY IN NEGATIVE FEEDBACK CONTROL OF INFLAMMATION
The innate immune system is characterized by exquisite sensitivity to markers of infection or tissue injury, very rapid production of powerful pleiotropic inflammatory mediators such as TNF, and a capacity for self amplification. These features create a danger of runaway inflammatory responses. Unprovoked, excessive or unduly prolonged expression of pro-inflammatory genes is normally prevented by multiple layers of negative feedback regulation. The p38 MAPK pathway is central to many of these feedback loops.
The p38 MAPK pathway regulates expression of DUSP1 via MSK-mediated phosphorylation of CREB/ATF transcription factors, which recognize highly conserved sites in the DUSP1 promoter [86-90]. The stability of DUSP1 mRNA has also been shown to be regulated by p38 MAPK [91] which is consistent with the identification of DUSP1 as a target of TTP [92-94]. Other studies implicate post-translational control of DUSP1 protein stability or catalytic activity by the p38 pathway [95, 96]. Whether transcriptional, post-transcriptional or post-translational mechanisms are involved, the control of DUSP1 function by p38 MAPK is an excellent example of negative feedback. But DUSP1 also regulates the activity of JNK and (under some circumstances) ERK. Therefore inhibition of p38 MAPK and blockade of DUSP1 expression or function might have unanticipated consequences, such as enhanced or prolonged activation of other signaling pathways.
The expression of TTP is controlled by the p38 MAPK pathway at several levels. Transcription is regulated by the MSKs [97], and stability of TTP mRNA is regulated by MK2 [98, 99]. The phosphorylation of TTP protein by MK2 causes its stabilization by preventing proteasome-mediated degradation [97, 98] (Fig. 3). The phosphate groups at serines 52 and 178 can be removed by protein phosphatase 2A (PP2A) [100], therefore the level of expression of TTP protein is tightly regulated by a dynamic equilibrium between MK2-mediated phosphorylation and PP2A-mediated dephosphorylation. At first sight it seems paradoxical that the p38 MAPK pathway promotes both the accumulation and the inactivation of TTP. This has been interpreted as a means of coupling the stability of pro-inflammatory mRNAs to the activation state of the p38 pathway. In the early phase of an inflammatory response, TTP is inactivated as it accumulates, allowing pro-inflammatory transcripts to remain stable for long enough to be translated. As cellular p38 activity declines, the equilibrium shifts in favour of TTP dephosphorylation and activation, so that the existing pro-inflammatory mRNAs can be rapidly degraded. A consequence of this intricate coupling is that, in vitro at least, long-term inhibition of p38 MAPK prevents the accumulation of TTP and renders target transcripts constitutively stable [48].
The cytokine IL-10 has complex, broadly anti-inflammatory effects on a wide variety of cells including macrophages [101-103]. In macrophages the expression of IL-10 is upregulated by pro-inflammatory stimuli, usually in a rather delayed manner. This can be seen as a homeostatic mechanism to guard against excessive inflammation. For example, experimental arthritis in the mouse can be treated using recombinant IL-10 [104], and is exacerbated in mice lacking IL-10 [105]. The interesting point here is that IL-10 expression is strongly regulated by p38 MAPK at both transcriptional and post-transcriptional levels. Transcription is upregulated by MSKs via phosphorylation of CREB/ATF proteins [87, 90]. p38-mediated phosphorylation of Sp1 has also been implicated in the control of expression [106, 107]. IL-10 mRNA is also recognized by TTP, and dramatically destabilized by inhibition of p38 MAPK [48, 108]. This nicely illustrates a general point about negative feedback effectors. Their expression is commonly controlled by the same signaling pathways that regulate expression of pro-inflammatory genes. Such sharing of control mechanisms will normally safeguard against unrestrained production of inflammatory mediators.
Another mechanism of negative feedback involves the p38-mediated phosphorylation of one or more of the TAK1-binding proteins, causing inhibition of TAK1 catalytic activity [109-112]. Inhibitors of p38 MAPK are thought to increase the activation of JNK and NF-κB by interfering with this negative feedback mechanism and thereby enhancing or prolonging TAK1 activation. MAPK p38 has also been reported to inhibit JNK signaling via effects on the upstream activator mixed lineage kinase 3 (MLK3) [113]. In addition, p38 MAPK may negatively regulate responses to IL-6 by increasing the expression of SOCS-3 (suppressor of cytokine signaling 3) [114-116]. There are likely to be several additional mechanisms of negative feedback regulation of the p38 MAPK pathway, or negative crosstalk between p38 MAPK and other pro-inflammatory signaling pathways.
THE P38 MAPK PATHWAY IN RHEUMATOID ARTHRITIS
The first generation of p38 MAPK inhibitors were discovered as compounds that decreased the expression of pro-inflammatory cytokines in myeloid cells [117]. Successive generations of increasingly potent and specific inhibitors have been invaluable tools for research into the biological roles of p38 α and β (whilst leaving the functions of p38 γ and δ more obscure). Information from many of these studies has added weight to the argument that chronic inflammatory diseases might be treated by targeting p38 MAPK [118-121].
Of the p38 MAPK family members it is almost certainly p38α that contributes most to the pathology of inflammatory arthritides. Experimental arthritis is unaltered in mice lacking p38β [122], whereas deletion in adult mice of the Mapk14 gene that encodes p38α protects them against TNF-induced inflammatory bone loss [123]. MK2 likely plays a critical role downstream of p38α , because the MK2 knockout mouse is protected against experimental arthritis [124]. Disruption of the genes encoding either MKK3 or MKK6 also protects against experimental arthritis [125, 126]. Upstream of this level there is not yet any definitive information. TAK1 protein and mRNA are expressed in RA synovial fibroblasts [127], but the mouse knockout is embryonic lethal. Perhaps conditional knockouts of the Map3k7 gene (which encodes TAK1) will reveal whether this kinase has an essential role in inflammatory arthritis, as suspected.
In the inflamed joints of patients with rheumatoid arthritis and in animal models of the disease, activation of p38 MAPK and the upstream kinases MKK3 and MKK6 is detected in macrophages and fibroblasts of the synovial lining layer, and at sites of bone erosion [128-134]. Activation of the p38α isoform appears to predominate, but there is also evidence of phosphorylated p38β and γ in certain cells [129]. Pro-inflammatory cytokines such as TNF and IL-1 in the inflamed joint presumably contribute to p38 MAPK activation. There may also be activation of the pathway by ligands generated locally through cell damage or proteolysis of the extracellular matrix.
As outlined above, p38 MAPK is believed to contribute to the overexpression of pro-inflammatory cytokines, chemokines, matrix metallo-proteinases and signaling enzymes such as COX-2 in the inflamed synovium. Phosphorylated p38 is also detected in the vascular endothelial cells of the blood vessels infiltrating the synovium, where it regulates expression of genes encoding cell adhesion molecules. Receptor Activator of NF-κB (RANK) plays a critical role in the differentiation and activation of osteoclasts, and therefore in the inflammatory osteolysis that accompanies RA [135, 136]. Engagement of RANK by its ligand RANKL activates p38, which in turn increases the expression and/or activity of transcription factors including NFAT2 (nuclear factor of activated T cells 2), MITF (microphthalmia-associated transcription factor) and PU.1 (purine-rich element binding factor 1) [137-144]. These transcription factors promote expression of cathepsin K, tartrate-resistant acid phosphatase and other genes essential to the resorptive function of osteoclasts. In chondrocytes the p38 MAPK pathway has been implicated in the expression of matrix metalloproteinase 13 (collagenase 3), which contributes to the degradation of cartilage [145-148]. Hence the fingerprints of p38 are clearly seen on several of the features of RA; abundant expression of pro-inflammatory cytokines and chemokines, increased vascularization and recruitment of lymphocytes, proteolytic degradation of bone and cartilage.
THERAPEUTIC TARGETING OF THE P38 MAPK PATHWAY IN RA
In a potentially crowded market, some investigators and pharmaceutical companies have chosen to focus on targets within the p38 pathway, other than p38 MAPK itself. For example, it has been argued that blocking the pathway at a higher level is likely to exert broader anti-inflammatory effects and reduce the risk of signal diversion into other pro-inflammatory pathways [149]. From this perspective TAK1 is an interesting candidate, since it appears to be involved in signaling downstream of several important receptors [120, 150-152]. However, involvement of TAK1 in the differentiation of regulatory T cells suggests that prolonged TAK1 inhibition might risk inducing or exacerbating autoimmune disease [153]. Another possibility is that therapeutic targets may be found amongst K63 ubiquitin ligases that assist in the initiation of p38 MAPK signaling [154]. At present, inhibitors of E3 ubiquitin ligases, TAK1, other MAP3Ks or even MAP2Ks are some distance from the clinic. A knockout mouse lacking the downstream kinase MK2 is viable and healthy but underexpresses pro-inflammatory cytokines and is relatively resistant to challenges such as LPS injection or experimental induction of arthritis [124]. This suggests that long term inhibition of MK2 may be a safe and effective means of treating RA and other diseases [151, 155]. A novel MK2 inhibitor was recently reported to inhibit paw swelling in the streptococcal cell wall model of arthritis [156]. However, some notes of caution need to be sounded. In one study the absence of MK2 exacerbated rather than ameliorated TNF-driven inflammatory bowel disease [157]. In a second, MK2 knockout provided no protection in some models of dermal contact hypersensitivity [158]. In another, MK2 mice were found to have increased susceptibility to the intracellular pathogen Listeria monocytogenes [159]. These findings point to certain concerns that might arise from therapeutic targeting of MK2.
In this field, the majority of pharmaceutical companies have invested in the development of specific inhibitors of p38 MAPK itself. Although several clinical trials have been completed, relatively little information has entered the public domain [160]. Yet, from available data, certain common themes are evident [149, 161]. In spite of increasing specificity of inhibitors, problems of toxicity in liver, skin and/or central nervous system have not been eliminated altogether. Because the inhibitors that have been tested belong to several distinct chemical classes, these adverse events could be mechanism-related, in other words unavoidable consequences of interrupting necessary physiological functions of p38 MAPK [2]. It is worth noting that observations from knockout mice suggest essential roles of the p38 MAPK pathway in muscle differentiation, erythropoiesis and bone formation [2, 162-164]. These observations raise concerns about p38 inhibition that remain (so far as we are aware) hypothetical.
More interestingly from the perspective of this review, a number of inhibitors displayed transient efficacy (for example as measured by down regulation of C-reactive protein). As one commentator has remarked, “The etiology of this phenomenon remains unknown but must lie in one or more biologic adaptations that allow escape from this pathway… One can’t help believing that to best understand the failure of this class of therapies, we need to better understand this biologic adaptation” [161].
It is likely that the failure of p38 inhbitors is connected to the important role of the p38 pathway in the negative feedback control of inflammatory responses. For example, as has been demonstrated in isolated macrophages, chronic inhibition of p38 may prevent expression of TTP, so that any pro-inflammatory mRNAs that are synthesised remain constitutively stable [48]. Because of extensive crosstalk, there is also the possibility that prolonged p38 inhibition may enhance the activity of other signaling pathways such as the JNK or NF-κB pathways, providing alternative routes to the expression of inflammatory mediators. Crosstalk between the p38 and JNK pathways may also provide an explanation for hepatoxicity in trials of p38 inhibitors. At any rate, enthusiasm for p38 inhibition now seems to be waning [118, 161]. For example, at the time of writing the website www.clinicaltrials.gov lists no ongoing trials of p38 inhibitors in RA. It may now prove difficult to identify the reason or reasons behind loss of anti-inflammatory efficacy, which is doubly unfortunate. The negative feedback loops that limit pro-inflammatory signaling pathways may provide a key to understanding not only the disappointing outcomes of p38 inhibitor trials, but also the chronicity of diseases like RA (see schematic figure in [165]).
Under most experimental conditions, negative feedback mechanisms ensure that p38 activation in response to a pro-inflammatory agonist is transient. In many cases the response is also followed by a refractory condition, in which the kinase pathway responds weakly to the same agonist or a different pro-inflammatory agonist (tolerance or cross-tolerance). Whilst this phenomenon is easy to study in static cell populations in vitro, the situation in vivo is much more complex, with interactions between distinct populations evolving as cells migrate into and out of sites of inflammation, proliferating, differentiating or dying as they do so. Nevertheless, there is a puzzle in the chronic activation of p38 MAPK that is observed in the rheumatoid joint. If negative feedback loops act efficiently to attenuate the response of the p38 pathway in individual cells, how can the pervasive activation of p38 in synovial tissue be explained? It has been suggested that RA depends upon a vicious cycle of inflammation, which provokes tissue damage and catabolism of extracellular matrix, generating ligands that activate Toll-like receptors, inducing inflammation … and so on. Persistence of inflammatory stimulus can help to explain pervasive p38 activation only if the refractory state is relatively short-lived, or if cell populations are continually replenished and it is the newly arrived cells, encountering the stimulus for the first time, that stain positive for activated p38 MAPK. But more than 75% of synovial lining cells and vascular endothelial cells in the inflamed synovium stain positively for phosphorylated p38, which is very difficult to explain in terms of activated incomer cells. In particular, vascular endothelial cells and fibroblasts are not thought to turn over rapidly, so the majority of activated cells of these types have likely been resident in the inflammatory milieu for some time. This suggests that negative feedback loops, which normally limit the strength and duration of p38 activation, are ineffective in the rheumatoid joint. In turn this raises new questions and opens new avenues for future research. Is there concrete evidence for failure of specific negative feedback mechanisms in chronically inflamed tissues? How might such apparently robust control systems break down, and could anything be done to prevent or reverse that malfunction, reinforce negative feedback and restore homeostasis? These questions might provide fertile ground for new approaches to the treatment of RA and other chronic immune-mediated inflammatory diseases.
ACKNOWLEDGEMENTS
Our work is supported by Arthritis Research UK, the Medical Research Council UK and the Kennedy Trust.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.

