All published articles of this journal are available on ScienceDirect.

Synovial Tissue Response to Treatment with TNF Blockers in Peripheral Spondyloarthritis
Abstract
This review describes the synovial response to treatment in peripheral spondyloarthritis (SpA). A series of recent studies demonstrates that the synovial histopathology is largely homogenous between different SpA subtypes and can be strongly modulated by effective treatment such as tumor necrosis factor (TNF) blockade. This includes a dramatic reduction of the infiltration with inflammatory cells (with the intriguing exception of B lymphocytes and plasma cells), a modulation of structural features such as vascularity, intimal lining layer hyperplasia, and ectopic lymphoid neogenesis, and a down-regulation of a variety of mediators involved in tissue damage. The analysis of tissue response to targeted therapies appears to be a novel and elegant approach to study the immunopathology of human peripheral SpA in vivo. Moreover, detailed cellular and molecular analysis of synovial features allows to identify synovial biomarkers of clinical response to therapeutic interventions which can be used in future early phase clinical trials in SpA.
INTRODUCTION
The successful introduction of tumor necrosis factor (TNF) blockade as a highly effective treatment for the different subtypes of spondyloarthritis (SpA) [1-4] has dramatically improved the clinical outcome and quality of life of SpA patients. An unforeseen consequence of this therapeutic success, however, is that clinical trial design in SpA is becoming increasingly challenging. On the one hand, the development of new treatment strategies is still necessary for the following reasons: 1) TNF blockade can be contraindicated in a subset of patients due to its safety profile, 2) a subset of treated patients experience an incomplete or non-response [5], 3) even in those patients with good clinical response, there is no convincing evidence yet that TNF blockade halts ankylosis [6, 7], 4) TNF blockade fails to induce a genuine sustained remission since almost all patients relapse shortly after treatment discontinuation [8].
On the other hand, the dramatic clinical efficacy of TNF blockade in a majority of patients questions the medical and ethical acceptability of large, long-term placebo-controlled trials in SpA. Therefore, it becomes increasingly important for new candidate drugs to ‘fail fast’, meaning that early phase proof-of-concept trials of limited size and treatment duration should lead to convincing ‘go/no go’ signals before proceeding with large long-term trials. Those drugs that fail in these proof-of-concept trials should not be used in larger patient populations, whereas obviously large scale phase IIb and III studies remain warranted to formally prove efficacy and safety for those drugs that make it through the proof-of-concept trials.
One way to address this new challenge is to develop sensitive biomarkers of treatment response. A biomarker is a characteristic that can be objectively measured and evaluated as an indicator of a normal biologic process, a pathophysiologic process, or a pharmacologic response to therapeutic intervention [9]. As such, changes in biomarkers during treatment indicate that this treatment has a genuine effect on a specific biologic process of interest. When such a marker additionally reflects a clinical outcome in a reliable, sensitive, and reproducible way, it can be evaluated as surrogate marker in individual patients. In the context of early proof-of-concept evaluation of new treatment modalities, one aims to see a clear biological effect at the group level rather than to predict individual clinical responses. Accordingly, we will focus here on biomarkers in this specific context and will not discuss their potential application as surrogate markers in clinical practice.
SYNOVIAL IMMUNOPATHOLOGY OF THE DIFFERENT SpA SUBTYPES
SpA has traditionally been subdivided in specific subsets (AS, reactive arthritis or ReA, psoriatic arthritis or PsA, inflammatory bowel disease (IBD)-associated arthritis, and undifferentiated SpA or USpA) according to the phenotypic presentation. This phenotypic classification reflects the fact that SpA can affect different tissues (synovium, enthesis and/or bone) in both axial and peripheral joints and is frequently associated with extra-articular manifestations in the gut (IBD), the skin (psoriasis), and the eye (acute anterior uveitis). More recently, the ASAS group is working on a new classification in axial SpA and peripheral SpA based on the observation that early forms of SpA often do not display yet all clinical features required for the classical phenotypic subclassification. The prototypical example is early axial SpA characterized by inflammatory low back pain which, however, can not be classified as AS because the radiologic sacroiliitis may only develop after several years of disease duration. Whereas this novel classification is expected to solve, at least partially, the issue of early disease, the main challenge remains to determine whether the different SpA subtypes and in particular the axial versus peripheral manifestations are driven by the same pathophysiological processes. Data of a recent cross-sectional inception cohort indicate that approximately 40% of the SpA patients have pure axial disease, 20% have exclusively peripheral joint involvement, and 40% have a combined phenotype with both axial and peripheral joint involvement (Paramarta et al, manuscript in preparation). In the context of biomarker research, the phenotypic diversity of SpA raises a double question: which biological compartment should be analyzed and are the pathophysiologic processes under investigation in this compartment similar in the different SpA subtypes?
As to the first question, it is important to notice that SpA is not a genuine systemic disease but rather a disorder which specifically affects distinct tissues exposed to mechanical or microbial stress. Accordingly, it is more likely that important pathophysiologic processes can be detected in the affected tissue than in peripheral blood. Of the affected tissues, the axial skeleton is the most frequent site of inflammation in AS as well as SpA as a whole but is extremely difficult to access. The studies of inflamed axial tissue are thus restricted to surgical samples or necropsy material [10, 11] and sequential sampling during treatment is technically impossible. In contrast, peripheral joint synovium, gut tissue, and skin can easily be accessed during active disease. As no studies have been performed on sequential gut biopsies in SpA and as tissue response in the skin in the context of PsA will be discussed in a separate chapter of this journal, we will focus here on peripheral joint synovium. Indeed, this tissue is an attractive target for biomarker research since more than half of the SpA patients and approximately one third of the AS patients have peripheral disease with preferential involvement of large joints such as ankles and knees, from which it is relatively easy to obtain sequential synovial tissue biopsies by needle arthroscopy [12]. As the synovial immunopathology is unaltered by the procedure or by ineffective therapy [13, 14], this approach allows to investigate the modulation of synovial immunopathology by novel treatments.
The second question is whether the synovial immunopathology is similar or not between different SpA subtypes. A number of early studies have compared the synovial histopathology of distinct SpA subtypes with rheumatoid arthritis (RA) (Fig. 1). The most consistent finding of these studies is a decreased synovial lining hyperplasia and an increased vascularity in both ReA and PsA in comparison with RA [15-20]. However, none of these studies directly compared different SpA subtypes. A recent systematic comparison between AS/USpA, PsA, and RA showed that the SpA subtypes shared the same immunopathologic features with significantly greater vascularity as well as infiltration with neutrophils and CD163+ macrophages compared to RA [21]. In contrast, lining layer thickness and the number of CD83+ dendritic cells were significantly increased in RA compared to AS/USpA as well as to PsA. In line with previous studies [22, 23], none of the SpA subtypes had positive staining for intracellular citrullinated proteins or major histocompatibility complex/human cartilage glycoprotein-39 complexes whereas >40% of the RA samples were positive for both markers. Moreover, there were no histological differences between oligo-articular and poly-articular PsA.
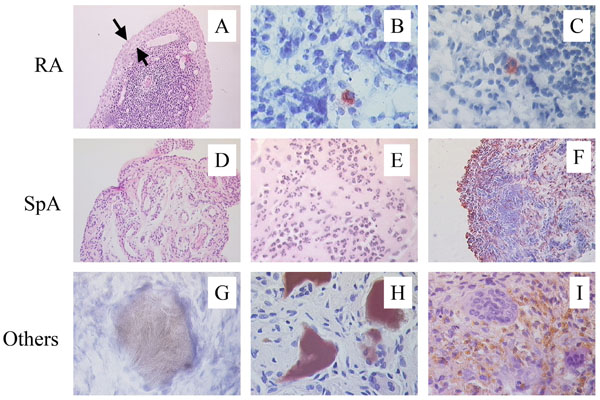
Distinct features of SpA synovitis. In comparison with RA, SpA synovitis displays less synovial lining layer hyperplasia (A), the abcens of intracellular citrullinated proteins (B), and the absence of HLA-DR4/HC gp-39 peptide complexes (C). Vascularity (D) as well as infiltration with polymorphonuclear cells (E) and CD163+ macrophages (F) is clearly increased in SpA. Depositions of uridic acid crystals (G), homogentisic acid (H), and hemosiderin (I) are typical for gout, ochronosis, and pigmented villonodular synovitis, respectively, and are not found in SpA synovitis.
The pathophysiologic similarities in synovial inflammation between different SpA subtypes and in particular between AS/USpA and PsA have recently been confirmed by the analysis of the inflammatory milieu in SpA and RA: the different SpA subtypes showed a consistent decrease in classically activated macrophage-derived mediators such as TNF and IL-1 in comparison with RA [24]. Coming back to the question which tissue should be sampled, it is also important to note that the alternatively activated CD163+ macrophages were not only increased in SpA synovium but also in gut biopsies of SpA and IBD patients [25, 26]. Although further research in potential synovial differences between SpA subtypes remains warranted, these data consistently indicate that the different SpA subtypes have largely similar synovial immunopathological characteristics which discriminates them from other inflammatory arthritides such as RA [27, 28].
MODULATION OF SYNOVIAL IMMUNOPATHOLOGY BY TNF BLOCKADE IN SpA
As there are no studies on synovial tissue response to treatment in AS solely but only in SpA as a group, and as the previously described synovial features correlate with disease activity across the different SpA subtypes [29], we will review the synovial response to treatment for the peripheral SpA group as a whole. Indeed, the only subset of SpA which has been studied as a separate entity is PsA as discussed in a different review article in this issue of the journal. Although in RA (and to a lesser extend in PsA) the synovial tissue response has been studied for a large array of disease modifying antirheumatic drugs (DMARDs) and biologicals [30-34], studies in SpA have only assessed the effect of TNF blockade with infliximab [14, 35] and etanercept [36]. This is partially related to the lack of efficacy of classical DMARDs, with the exception of sulphasalazine, as well as novel biologicals in peripheral SpA. It remains thus unclear whether the synovial changes observed with TNF blockers reflect specific mechanisms of action of this class of drugs or can be used more broadly as biomarkers of effective treatment in peripheral SpA.
MODULATION OF THE INFLAMMATORY CELL INFILTRATION
A first major observation is that the global inflammatory cell infiltration is significantly reduced upon treatment with TNF blockade. Studies with infliximab in patients with different SpA subtypes demonstrated that especially infiltrating polymorphonuclear (PMN) cells and MRP8+ monocytes were rapidly reduced, which was already the case 1 or 2 weeks after the first infusion [35]. This trend was further confirmed after 12 weeks of infliximab, when not only infiltrating monocytes and granulocytes, but also resident tissue CD163+ macrophages and T lymphocytes were significantly reduced [14, 35, 37]. Similar effects were observed with etanercept and the decrease in synovial infiltration was maintained or even augmented on the long term [36].
The decrease of the cellular infiltration upon TNF blockade could be due to a diminished influx, an increased local apoptosis, and/or an accelerated efflux of these cells. The fact that the number of infiltrating MRP8+ and MRP14+ monocytes is reduced more rapidly and more profoundly than the number of CD163+ resident tissue macrophages in the synovial tissue, together with the early and significant decrease of serum MRP8/MRP14 levels in contrast to the sCD163 levels, strongly suggest a primary effect on inflammatory cell recruitment into the inflamed synovial tissue by TNF blockade [37]. This hypothesis is further confirmed by the treatment-induced down-regulation of the number of synovial blood vessels as assessed by CD146 and von Willebrand Factor (vWF) immunostaining as well as the expression of markers of neovascularisation such as αVβ3 integrin, angiogenic factors such as synovial matrix metalloproteinase (MMP)-9, and adhesion molecules [14, 35, 36, 38, 39]. The reduction of hypervascularity and endothelial activation is likely to restrain the migration and homing of inflammatory cells into the synovium.
As for the contribution of local cell death, a couple of studies in Crohn’s disease have proposed that apoptosis of activated lymphocytes by outside-in signaling is likely to play a role in the immunomodulating effect of infliximab. The absence of this effect by etanercept could thus potentially explain the lack of efficacy of this drug in IBD. However, the successful use of certolizumab, a pegylated TNF blocker which does not induce cell death, in Crohn’s disease [40, 41] challenges this theory. As to arthritis, only one study in RA has reported a contribution of apoptosis to the synovial effects of TNF blockade [42]. In contrast with the IBD studies, this report suggests apoptosis of macrophages rather than lymphocytes and found this effect both with etanercept and infliximab. However, the large majority of studies in RA, PsA, as well as SpA could not provide evidence for an increased synovial cell death upon TNF blockade, making it unlikely that this mechanism plays a dominant role in the synovial tissue response [43-46].
A third and most intriguing hypothesis is that TNF blockade does not only reduce the influx but also promotes the efflux of inflammatory cells from the inflamed tissue. This theory is supported by a recent study in the TNF transgenic mouse model of arthritis showing an increase of synovial lymphatic vessels upon TNF blockade [47]. A similar increase of lymphatic vessels was observed in RA and SpA synovium upon infliximab treatment.
MODULATION OF SYNOVIAL B LYMPHOCYTES
In sharp contrast with the effect on neutrophils, macrophages, and T lymphocytes, the synovial infiltration with B cells and plasma cells was not reduced upon treatment and in a few cases the presence of plasma cells was even clearly augmented [14, 35-37]. Downregulation of the number of CD68+ macrophages and T cells with unchanged number of B cells and plasma cells was also observed in two other studies involving treatment with infliximab in patients with PsA [45, 48]. Whereas the exact biological meaning and the underlying mechanisms of these findings are still unclear, it suggests that TNF blockade does not affect or even promotes infiltration, survival, local proliferation, and/or differentiation of B cells but no other leucocytes. Interestingly, this phenomenon is completely uncoupled from clinical or histological inflammation and thus indicates that these B cells and plasma cells are probably not primary players in the synovial immunopathology in SpA [49]. This should also be interpreted in the context of other signs of de novo B cell activation upon treatment with infliximab such as the impressive induction of anti-nuclear antibodies (ANA) and IgM anti-dsDNA antibodies [50, 51] and warrants further investigation of B cell biology in SpA.
In this context, it is interesting to note that the microarchitectural organization of infiltrating B cells in ectopic lymphoid structures is not restricted to seropositive diseases such as RA but is equally found in seronegative joint inflammation including the different subtypes of SpA [52-55]. Accordingly, synovial ectopic lymphoid neogenesis is more related to the degree of local inflammation than to the presence of autoantibodies and is downregulated by TNF blockade in SpA.
MODULATION OF TLR EXPRESSION
It has been suggested that activation of innate immune cells in the gut and in the joints by microbial pathogens may be relevant to inflammation in SpA with a special role for the CD163+ macrophage subpopulation [25]. This has been linked to toll like receptors (TLRs). TLR-2 and TLR-4 bind to components of both Gram-positive and Gram-negative bacteria. Following ligand binding TLRs activate a signaling cascade which results in the production of mediators of inflammation. TLRs have been identified in RA synovium and have been shown to play an important functional role in experimental arthritis models. The synovial expression of both TLR-2 and TLR-4 is significantly higher in SpA than in RA despite similar levels of local inflammation [56]. Both infliximab and etanercept sharply down-regulate the synovial TLR expression. These findings highlight the potential role of innate immunity related inflammation in SpA and provide new insights into the mode of action of TNF blockade.
MODULATION OF STRUCTURAL DAMAGE
As the inflamed synovium plays a crucial role in structural damage to articular cartilage and bone, it is important to know whether TNF blockade only blocks inflammation or also affects mechanisms of joint destruction. The synovial intimal lining layer, consisting of both fibroblast-like synoviocytes (FLS) and macrophages, plays an important role in this process by promoting osteoclast differentiation and activation through the RANK-RANKL system, by production of MMPs which can degrade cartilage, and by a direct effect of the FLS-adhesion molecule cadherin-11 on cartilage [57].
TNF blockade appears to influence several of these factors in the inflamed SpA synovium. Firstly, TNF blockade reduces the degree of lining layer hyperplasia [14, 35, 36]. The thickness of the synovial lining was even normalized after long-term treatment. This effect is likely to be mediated by a reduction of TNF-induced proliferation and/or a restoration of Fas-mediated apoptotic cell death of synovial fibroblasts [58], since the number of CD55+ synovial lining fibroblasts were significantly reduced after treatment [35]. Secondly, both MMPs and their inhibitors, tissue inhibitor metalloproteinases (TIMPs), were not only abundantly expressed in SpA synovitis but were rapidly and profoundly downregulated by TNF blockade [39]. Thirdly, the expression of cadherin-11 by FLS appeared to be TNF-driven in vitro and, accordingly, was significantly downregulated by anti-TNF treatment in vivo [59]. All these effects may contribute to halt the structural damage as demonstrated by the absence of progression of radiological damage over a 2-years period of follow up [36].
In contrast with MMPs and cadherin-11, the abundant synovial expression of RANK, RANKL, and osteoprotegerin (OPG) in SpA synovitis appeared to be largely independent of the degree of synovial inflammation and was thus only downregulated by TNF blockade in the subset of patients with the largest systemic response [60]. However, this absence of effect on the RANK/RANKL/OPG system does not necessarily mean that TNF blockade does not affect osteoclast differentiation and activation as TNF itself has a direct role in this process [61]. Finally, it should be noted that the effect of TNF blockade on synovial mediators of new bone formation and ankylosis, such as the bone morphogenetic protein (BMP) and Wnt signaling pathways, remains largely unexplored in SpA.
CONCLUSIONS AND FUTURE DIRECTIONS
Taken together, these studies demonstrate that the synovial histopathology is largely homogenous between different peripheral SpA subtypes and can be strongly modulated by effective treatment such as TNF blockade. This includes a dramatic reduction of the infiltration with inflammatory cells (with the intriguing exception of B lymphocytes and plasma cells), a modulation of structural features such as vascularity, lining layer hyperplasia, and ectopic lymphoid neogenesis, and a down-regulation of a variety of mediators involved in tissue damage.
The analysis of tissue response to targeted therapies appears to be a novel and elegant approach to study the immunopathology of human disease in vivo. This is of particular importance in SpA as there is scarcity of appropriate experimental models for this disease. The use of such therapies allows to assess if such pathways are TNF-driven or not and whether specific cell types or molecules really contribute to the synovial inflammation. Although the studies performed until today are largely restricted to immunohistology, the translational combination of novel targeted therapies and more sophisticated molecular methods to analyze gene expression, T cell receptor (TCR) and B cell receptor (BCR) repertoires, … are likely to reveal fundamental disease mechanisms in the near future [62-64].
The discussed studies also raise the possibility to use synovial immunopathology as an additional biomarker to discriminate between effective and ineffective treatment in early phase clinical trials in SpA. In a proof-of-concept study, we demonstrated that changes in different macrophage subsets (CD163+ resident tissue macrophages and infiltrating MRP8+ or MRP14+ monocytes), PMN cells, and MMP-3 expression are superior to C reactive protein (CRP) to discriminate between TNF blockade and placebo treatment in SpA [65]. As not all SpA patients have peripheral arthritis and as it remains unclear to what extend peripheral and axial disease are driven by the same processes, it is important that several synovial markers (such as MMP-3 and MRP8/14) can easily and reliably be measured in serum [37, 39, 66]. This approach will not be restricted to markers of synovial inflammation as soluble biomarkers of cartilage and bone metabolism also appears to be modulated by TNF blockade in SpA [67, 68]. The main challenge will thus be to set up a panel of reliable markers reflecting the pivotal biologic processes of SpA pathogenesis (including enchondral bone formation) and to assess this panel across different targeted treatment strategies in both axial and peripheral SpA.
ACKNOWLEDGEMENT
None declared.
CONFLICT OF INTEREST
None declared.

