All published articles of this journal are available on ScienceDirect.

The Synovium in Rheumatoid Arthritis
Abstract
Rheumatoid arthritis (RA) is a chronic autoimmune disease targeting multiple joints. The synovium is the primary site of the inflammatory process, which if untreated leads to irreversible damage to the adjacent cartilage and bone. It is now well established that autoantibodies that are characteristic of RA, including rheumatoid factor (RF) and anti-citrulluninated protein antibodies (ACPA), are present before clinical disease onset. Studies in both humans and animal models are beginning to provide new insights into how this asymptomatic autoimmunity evolves into an inflammatory process that is localized in the synovium.
Once RA synovitis established, a number of amplification mechanisms serve to sustain the process leading to the persistence of the disease. These mechanisms include engagement of the resident mesenchymal cells and the establishment of ectopic lymphoid structures in the synovium, although the relationship between these lymphoid structures and the presence of RA autoantibodies remains unclear.
An enhanced understanding of the mechanisms that initiate and sustain RA synovitis offers unprecedented opportunities for therapeutics, and ultimately prevention strategies.
INTRODUCTION
The transition from normal to non-specific inflammatory synovium and subsequently to established rheumatoid arthritis synovium is the result of a break in immune tolerance both systemically and at the level of the synovial tissue. The presence of circulating ACPA and RF years prior to the onset of clinical symptoms [1, 2] and early histological changes in the (presumed) unaffected joints of RA patients [3, 4] indicates that the early stages of this immune tolerance breakdown are subclinical. The importance of mediators of innate immunity in initiating synovial responses is being increasingly recognized. Recent progress has been made in elucidating the mechanisms involved in bridging the innate and adaptive immune systems leading to persistent synovitis, however our understanding of these mechanisms is still incomplete. This review will describe the main histopathological and immunohistologic features of RA synovitis, and will address some potentially important mechanisms that may facilitate the initiation and progression of rheumatoid arthritis synovitis.
PRE-CLINICAL STAGE OF RA
Circulating autoantibodies to citrullinated proteins (ACPA) can be detected several years prior to the onset of joint symptoms [1, 2] and examination of pre-disease cohorts of patients at risk for future RA has suggested a progression in the fine specificity of these responses consistent with epitope spreading [5]. This phenomenon has also been demonstrated in other autoimmune disorders such as systemic lupus erythematosus [6]. Citrullination itself is not unique to RA tissues, and indeed is a normal physiological process. While the source of the citrullinated protein autoantigens in established RA appears to be in the synovium [7, 8], extra-articular sites such as the lungs [9], or skin [10], are also potential sources. Isoforms of peptidylarginine deiminase (PAD), the enzyme responsible for post-translational conversion of arginine to citrulline in peptides, are found in a variety of cells and tissues [11-17], however to date only PAD2 and PAD4 have been found in synovium [7, 18]. PAD4 expression seems to be more specific for RA tissue, however the expression of both PAD2 and PAD 4 correlate with the degree of inflammation. Thus the development of immune responses to citrullinated proteins as indicated by the presence of ACPAs is highly suggestive of a break in immune tolerance that precedes the onset of clinically detectable synovitis.
NON-SPECIFIC SYNOVITIS, IMMUNE COMPLEXES AND INNATE IMMUNITY
Studies of the earliest stages of synovitis have mostly relied on animal models of rheumatoid-like inflammatory arthritis and although the direct application/attribution to human disease should be interpreted with some caution, these studies provide valuable insights into early pathogenesis. In the G6PI (KBxN) immune transfer mouse model of rheumatoid arthritis, components of the innate immune system including mast cells [19, 20], complement [21], Fc receptors [22], and cytokines such as IL-1 and TNFα [23] are critical for the development of synovitis. In this model, the formation of immune complexes appears to be an important initial step to allow pathogenic autoantibodies access to the synovium [24]. FCRγ-expressing cells in inflamed synovium may sense these circulating immune complexes leading to an increased vasopermeability preferentially in the distal extremities. On this basis, it is proposed that distal extremity vessels may have unique properties and that the increased vasopermeability may also have an amplifying function in the early stages of synovitis allowing influx of inflammatory cells into the joint tissues. It is well known that immune complexes are present in blood and synovial fluid of patients with established RA, but their role in the early stages of the disease is less clear. It has been shown that in a significant proportion of ACPA positive RA patients these circulating immune complexes contain citrullinated fibrinogen [25]. Immune complexes and citrullinated fibrinogen co-localize with complement component C3 in pannus tissue suggesting immune complex deposition and complement activation may lead to ongoing synovitis [25]. Recently, Monach et al. demonstrated that immune complexes deposited in synovium target a wide range of proteins some of which are histones and relatively specific for RA [26].
The potential importance of immune complexes in initiating human disease is supported by an illustrative case report which describes the progression of an initially asymptomatic individual who was ACPA positive, and who subsequently developed synovitis [27]. Although anti-CCP antibodies preceded joint symptoms, the development of antibodies to citrullinated fibrinogen coincided with the appearance of joint symptoms. On synovial biopsy of an affected knee performed soon after symptom onset, the lining layer was markedly abnormal whereas the sublining layer showed a histologically normal vascular pattern, and minimal if any increased cellularity. In the lining layer there was evidence of immune deposits of IgG, IgA, C3, and fibrin which co-localized with intra-cellular citrullinated proteins, and with widespread lining cell apoptosis (Fig. 1). This supports the hypothesis that pre-existing ACPA may access synovial tissue early in disease perhaps at the time of non-specific synovitis onset and that the synovial lining layer may be the primary target of the initial immune processes. It can be further speculated that the immune mechanisms generating ACPA are then amplified with continued exposure to citrullinated synovial antigens. In a similar case series of very early arthritis with a mean disease duration less than 4 weeks, perivascular CD3 and CD4 T cells and a mild superficial infiltration of lymphocytes and macrophages were found, however there were no aggregates and vascularity was normal, again suggesting superficial structures such as the lining layer may be a primary site for disease pathogenesis [28].

Synovial tissue analysis of biopsy material obtained from a symptomatic knee joint shortly after onset of articular symptoms. A) H&E light microscopy showing marked disruption of the synovial lining cell layer. The lining cells appear to be floating in an amorphous extra-cellular matrix. The sublining stroma is relatively unremarkable with minimal evidence of inflammatory infiltration. B) Intracellular citrullinated antigens are detected by intense staining of the synovial lining cells using a polyclonal anti-citrulline antibody. There is less intense staining of the surrounding matrix. C) Immunofluoresence staining of synovial tissue for C3 showing positive staining in the lining layer. Similar results were seen with IgG, IgA, and fibrin staining. D) The synovial lining cells exhibit evidence for widespread apoptosis as detected by TUNEL staining. Original magnification x 200 for A), and x 400 for B), C), D).
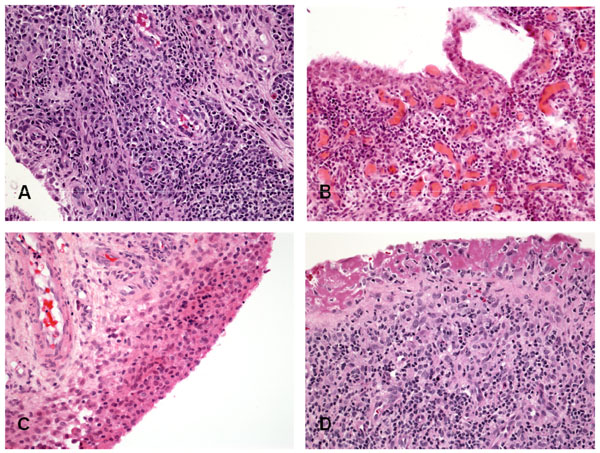
Histopathology of RA synovitis. (A) lymphoid aggregate; (B) Diffuse lymphocytes infiltrate; (C) Hyperplasia of the lining layer; (D) Fibrin cap replacing a denuded lining layer.
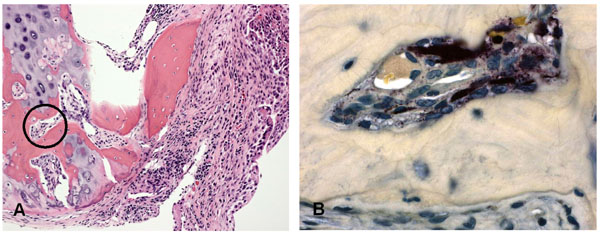
Interface between pannus tissue and bone in a patient with RA. (A) the synovial lesion is invading the adjacent bone. (B) Staining for tartrate resistant acid phosphatase in the circled area demonstrates the presence of osteoclasts.
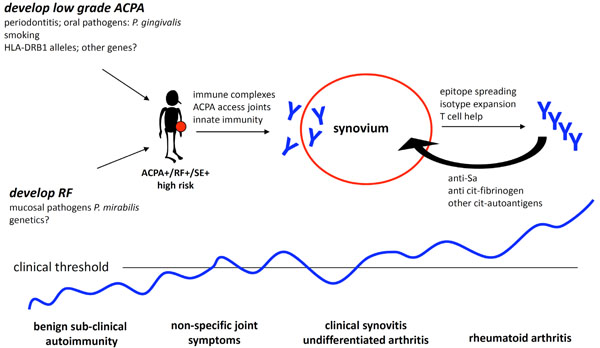
Conceptual framework for the evolution of RA synovitis.
DENDRITIC CELLS AND INNATE IMMUNITY IN RA SYNOVITIS
Dendritic cells (DC) are key players in innate immunity and may initiate and/or perpetuate synovitis by presenting arthritogenic antigens to components of the adaptive immune system [29-31]. Activation and maturation of DC occurs after exposure to multiple triggers including viral and bacterial products, cytokines, immune complexes, multiple endogenous ligands, and even disruption of cell-cell contact [32] and hypoxia [33]. Myeloid DC (mDC), and the more immature plasmacytoid DC (pDC) express distinct patterns of chemokine receptors and Toll-like receptors (TLR) [29, 34]. Both mDC and pDC are present in inflamed synovial tissue, and are not specific to RA [35-37]. However the number of pDC is higher in RA synovium compared to OA synovium, particularly in synovium from patients who are RF or ACPA positive. This observation is consistent with the suggestion that pDC may be involved in the synovial immune responses leading to the local formation of RA autoantibodies. In RA synovium, DC are located in primarily in the sublining tissues near aggregates of CD3 and CD8 cells [36], and adjacent to blood vessels [35]. Synovial pDC retain an immature phenotype and express IL-18 and IL-15, which may contribute to synovial inflammation and further DC recruitment, whereas synovial mDC produce IL-23 which may promote Th17 cell expansion [36]. The proportion of mature mDC increases with increasing grade of inflammation [37], possibly reflecting ongoing recruitment and activation of DC in persistent disease.
FIBROBLAST-LIKE SYNOVIOCYTES AS MEDIATORS OF INNATE IMMUNITY IN RA SYNOVITIS
RA fibroblast-like synoviocytes (FLS) express several TLR, suggesting they may contribute to the innate immune responses initiating and/or perpetuating RA synovitis. TLR3 and TLR4 in particular are highly expressed in RA synovium, even at an early stage of disease, and are found predominantly in the lining layer, occasionally extending into the sublining and perivascular areas [38]. TLR3 stimulation of RA FLS leads to increased expression of inflammatory cytokines such as IL-6, and to matrix degrading enzymes such as MMP3 and MMP13 [38]. A number of endogenous ligands stimulate TLR pathways in RA FLS [39], and can thus serve to activate these cells.
TRANSITION FROM EARLY NON-SPECIFIC SYNOVITIS TO ESTABLISHED RA SYNOVITIS
Key histological features of RA including synovial lining layer hypertrophy, sublining infiltration with mononuclear cells, increased vascularity and fibrin deposition are seen in patients reporting as little as 6 weeks or less of symptoms and in the unaffected joints of patients with active synovitis [3, 4], whereas organized lymphoid follicles and pannus tissue are features generally associated with established RA synovium (Fig. 2). As previously described, abnormalities of the synovial lining layer may be a very early feature of synovitis. Even in early clinical disease, the surface of the lining layer is often covered with fibrin deposits generated from the activation of the fibrinolytic system by synovial fluid. Indeed, the lining layer may be completely replaced by a fibrin cap, and in highly inflamed tissues this fibrin can extend deep into the sublining layers. In very early disease the sublining infiltrates may be minimal or modest [27, 28]. At this stage, macrophages [3], and an infiltration by natural killer cells has been described [40]. Diffuse mononuclear infiltrates and small lymphoid aggregates are seen in both early and late RA. However, as discussed in detail below, aggregates resembling lymphoid follicles with germinal centers are typically seen only in well established disease. The synovium obtained from ACPA positive RA patients tends to have more ectopic germinal centers and reduced fibrosis compared to synovium from ACPA negative patients, regardless of disease duration [41, 42], although other studies could not confirm these observations [43-45]. Pannus, the highly destructive tissue present at the interface between synovium, cartilage, and bone is a characteristic feature of erosive RA and contains large numbers of macrophages and fibroblasts that express high levels of proteases. Osteoclasts derived from pannus tissue can be detected at the interface of bone and cartilage (Fig. 3).
Vascular patterns are also established early in disease and may serve to distinguish RA from other non-RA arthropathies [46, 47]. In established RA, vascular structures are prominent especially in the deep sublining areas presumably related to increased angiogenesis. Immature blood vessels can be found early in disease but are even more numerous in patients with erosive disease of longer duration indicating ongoing angiogenesis [48]. Despite evidence of angiogenesis, morphometric studies indicate there are relatively fewer blood vessels in sites adjacent to the expanded lining layer which when combined with the increased metabolic demands of the highly cellular structures, suggests relative hypoxia [49]. Hypoxia is a potent stimulus for the production of VEGF and other angiogenesis mediators which are highly expressed in inflamed RA synovium [50-52]. The vascular endothelium is activated by pro-inflammatory mediators to express adhesion molecules such as E-selectin, P-selectin, and ICAM1 that are involved in the recruitment of inflammatory cells. Synoviocytes also express adhesion molecules including VCAM1, VLA4, PECAM1 and ICAM1. These molecules are expressed equally in synovium from patients with early (<1 year symptoms) and late RA [53]. It is likely that adhesion molecules play a role in initiating and maintaining synovial infiltrates by recruiting inflammatory cells through the endothelium and then retaining inflammatory cells in the synovium through adherence to matrix cells and other synovial structures.
Histological comparisons of synovial tissue obtained from patients that have been matched for disease activity and treatment have shown no difference in cellular infiltration between those with early (<1 year) or late (>5 years) disease [54, 55]. Similarly, cytokine expression [56] and adhesion molecule expression [53] is similar between early and late disease. This supports the hypothesis that the earliest synovial pathology is subclinical, the duration of subclinical disease may be variable and that at the time of symptom onset, clinical assessment and subsequent tissue biopsy, the synovitis is already in a chronic stage [57]. In the absence of serial sampling of joints that progress from asymptomatic to minimally symptomatic to definite clinical synovitis to erosive synovitis it is difficult to assess the true histological progression of disease. Such studies are difficult to perform.
ECTOPIC LYMPHOID NEOGENESIS IN RA SYNOVIUM
One of the important unanswered questions in RA pathogenesis is how the systemic autoimmunity that precedes the clinical onset of RA becomes localized and amplified in the joints. To address this key question, a number of studies have aimed to develop a better understanding of ectopic lymphoid neogenesis, a process whereby lymphoid structures resembling those present in secondary lymphoid tissues become established in target tissues such as RA synovium. Research in this area has generated a number of important observations:
- Fully developed lymphoid aggregates with functional germinal centres are found in ~25% of RA synovial tissue samples, typically those derived from individuals with well established disease [44, 45, 58-62]. One hypothesis has suggested that the capacity to form these ectopic lymphoid structures in the synovium is an inherent property of the disease process in a subset of RA patients and one that is retained throughout the duration of the disease [60, 62]. Other viewpoints suggest that these structures are a non-specific reflection of the chronicity and severity of the disease, are not indicative of a unique immunopathologic process in a subset of RA patients [44, 45, 61] and may be reversible [43]. It should be added that ectopic lymphoid structures are by no means unique to rheumatoid synovitis, and have been well described in a spectrum of other chronic autoimmune diseases.
- Several key cytokines and chemokines have been shown to be involved in the formation of ectopic lymphoid structures in the synovium. These include CXCL13, CCL21, LTα,β [62-65]. The chemokine CXCL13 has been of particular interest since it is important in the recruitment of B cells, and was shown to be actively produced by antigen experienced synovial helper T cells [64].
- A recent detailed study of RA lymphoid aggregates indicates that synovial tissues with follicular structures characterized by the presence of follicular dendritic cell (FDC) networks express activation-induced cytidine deaminase (AID), the enzyme required for somatic hypermutation and class-switch recombination of Ig genes [59]. These structures were found to be spatially associated with plasma cells that produce ACPA. Moreover, elegant experiments where human RA synovium was transplanted into SCID mice provided compelling evidence that these AID positive FDC structures support ongoing production of class-switched ACPA which were detectable in the sera of the mice, without any requirement for further recruitment of immune cells, particularly B cells, into the lymphoid structures. While this study provides evidence that ectopic lymphoid structures in the RA synovium are functional and support autoantibody maturation and production, several other large studies have failed to demonstrate an association between ectopic lymphoid structures in the synovium and B cell autoimmunity in RA [43-45]. In particular, ectopic lymphoid neogenesis is described in RF and ACPA negative RA [43], and in psoriatic arthritis [58]. Thus, it remains a challenge to clearly define the pathogenic significance of these synovial lesions, which are characteristic although not specific for RA.
INTERACTIONS BETWEEN IMMUNE CELLS AND RESIDENT MESENCHYMAL CELLS IN THE SYNOVIUM
Irrespective of how the mononuclear cell infiltrate is arranged in the synovium, be it diffuse, aggregated, or with germinal centre formation, there is clearly close interaction between immune competent cells such as lymphocytes, macrophages, and DC with resident mesenchymal cells of the synovium such as FLS. The interactions between these cells are meditated by complex cytokine networks and by signaling events resulting from direct cell to cell contact. Much has been written about the former, as the study of cytokine effects on various cell types lends itself well to in vitro manipulation and experimentation. For example, the effects of key pro-inflammatory cytokines such as TNFa and IL1b have been exhaustively studied and exploited therapeutically. More recently, the recognition of the key pro-inflammatory role that the T cell cytokine IL-17 plays in RA synovitis, has generated similar investigative activity to better define the effects of this cytokine on cells in chronic inflammatory microenvironments such as the RA synovium (reviewed in [66, 67]).
In contrast to defining the effects of cytokines such as TNFa, IL-1b, and IL-17 on cell biology, the effects that are mediated by cell-cell contact are more subtle, and more difficult manipulate in vitro. The close in vivo juxtaposition of T cells, B cells, macrophages, dendritic cells, and FLS in the synovial microenvironment suggests that the signaling events mediated by direct cell-cell contact between these cells likely involve multiple partners simultaneously, a situation that is difficult to simulate in vitro. Nevertheless, an interesting recent study demonstrated that in vivo activated CD14+ monocytes derived from the inflamed joints of patients with active RA spontaneously and specifically promoted Th17, but not Th1 or Th2 responses, compared with resting CD14+ monocytes from the blood. These in vivo activated monocytes promoted Th17 responses in a cell-contact dependent manner, and unlike Th17 stimulation by monocytes that were in vitro activated with lipopolysaccharide, intracellular IL-17 expression was TNF-alpha- and IL-1beta-independent. These data suggest that newly recruited memory CD4 T cells can be induced to produce IL-17 after cell-cell interactions with activated monocytes in the synovium [68]. This mechanism may be particularly relevant in explaining the incomplete responses observed with TNFa inhibition and the recurrence of disease with withdrawal of therapy.
The interactions between immune/inflammatory cells and FLS are of particular interest since they are an important link between the inflammatory and destructive arms of RA synovitis. RA T cells, which have been shown to exhibit extensive features of premature senescence [69], aberrantly express the fractalkine (FKN) receptor CX3CR1 which interacts with FKN expressed on FLS. This interaction results in activation and enhanced survival of the T cells [70], as well as proliferation of the FLS [71]. A parallel interaction is mediated through the aberrant expression of natural-killer group 2, member D (NKG2D) and its ligand MIC expressed on FLS [72]. Such self-sustaining amplification loops between synovial T cells and FLS potentially plays a key role in the perpetuation of RA synovitis, while increasing its destructive potential.
Much has been learned over the past two decades about the biology of FLS and how they become “transformed” in the RA synovial microenvironment to become efficient matrix degrading cells that make a major contribution to the articular damage seen in RA joints. An important recent insight has been the identification of cadherin 11 as the adhesion molecule that mediates the homotypic aggregation of FLS in the synovial lining layer [73-75]. These studies have clearly shown that cadherin 11 is an important mediator of RA FLS invasiveness, and that inhibition of this molecule results in a dramatic amelioration to the articular damage caused by chronic synovitis. Additionally, SCID mouse studies have demonstrated the ability of human RA FLS to migrate and damage distal unaffected human cartilage in a process that may be facilitated by angiogenesis. Therapeutic exploitation of these observations may add substantially to the current armamentarium, which is focused primarily on inhibiting inflammatory mediators and cells rather than matrix degrading FLS.
CONCLUSIONS
Recent insights obtained from animal models and studies of early and established RA synovial tissues have suggested potential mechanisms by which normal immune tolerance is interrupted leading to autoimmunity (Fig. 4). A non-specific insult could potentially trigger local innate immunity heralding the onset of clinical synovitis. In the setting of autoimmunity this inflammation persists leading to the formation of organized lymphoid structures and angiogenesis which serve to sustain the inflammatory synovium. In time, mesenchymal transformation and osteoclastogenesis lead to the destructive lesions characteristic of established RA. This paradigm suggests steps at which targeted intervention has the potential to significantly alter the course of disease and identifies structures and mechanisms which may be predictive of disease outcome or treatment response.
ACKNOWLEDGEMENT
None declared.
CONFLICT OF INTEREST
None declared.

