All published articles of this journal are available on ScienceDirect.

Long Term Effects of Cyclophosphamide Treatment on Lung Function and Survival in Scleroderma Patients with Interstitial Lung Disease
Authors Info & Affiliations
Abstract
Background:
Scleroderma (SSc) patients with active interstitial lung disease (ILD) experience a decline in lung function and increased mortality; cyclophosphamide (CYC) therapy may stabilize lung function at one and two years follow-up. Long-term lung function and survival outcomes of SSc patients with ILD following CYC treatment remain largely unknown.
Methodology:
We reviewed records of SSc patients with active ILD who had received at least six months of CYC treatment and had pulmonary function tests (PFTs) performed at least two years from the onset of treatment.
Principal Findings:
Thirty eight patients meeting eligibility criteria had a mean follow-up period from start of CYC to the last follow-up PFT of 5.1 years (range 2.3 -10.8 years). At a median of 4.1 years (range 9 months - 8.4 years), 12/38 (32%) patients had a significant decline in % predicted Forced Vital Capacity from their baseline PFT. At a median of 3.9 years (range 7 months - 8.4 years); 12/36 (33%) patients experienced a significant decline in their % predicted carbon monoxide diffusing capacity. Three patients died at a follow-up between 4.5-6 years and two received bilateral lung transplants because of severe restrictive lung disease.
Conclusions:
While the majority of SSc patients treated with CYC for active ILD experience long-term lung function stability and survive, greater than 1/3 of patients will experience either lung function decline, death, or require a lung transplant. This suggests that despite aggressive immune suppressing therapy, a subset of patients will have continued lung function decline, highlighting the need for ongoing monitoring and better therapeutic options.
INTRODUCTION
Interstitial lung disease (ILD) is one of the leading causes of death among patients with systemic scleroderma (SSc) due to the fact that a significant subset of SSc patients will have progressive and severe restrictive lung disease [1, 2]. Two prospective, double-blind, randomized controlled trials have demonstrated that cyclophosphamide (CYC) stabilizes lung function in SSc patients with ILD [3-5]. While CYC’s effect on lung function is known up to a follow-up period of 2 years, little is known about the sustainability of lung function response long term. Little is also understood about CYC’s effect on survival [3, 6]. This retrospective study was designed to determine long term lung function and survival in SSc patients with ILD who were treated with a course of CYC treatment and had more than 2 years of follow up data.
MATERIAL AND METHODS
Study Population
The Johns Hopkins Scleroderma Center database was screened for eligible SSc patients ever treated with CYC and seen in clinic from March 27, 1991 to April 30, 2006. Since medication data was routinely entered in the database after 2002, records of scleroderma patients who had a bronchoalveolar lavage (BAL) for evaluation of ILD prior to 2002 were screened to identify patients treated with CYC, as it was standard practice in our center to perform BAL for evaluation for possible alveolitis [6-8].
SSc patients with ILD treated with CYC were included if they met the following criteria: [1] had at least 6 months of CYC treatment [2], had been followed for at least 2 years from onset of CYC therapy, and [3] had a follow-up PFT more than 2 years from the PFT date at ILD diagnosis. The diagnosis of ILD was made clinically by the treating clinician; patients had to have at least one of the following: [1] pulmonary fibrosis with or without ground glass opacities on chest x-ray or computed tomography scan [2], an abnormal PFT (either percent predicted FVC or DLco <80%), or [3] an abnormal bronchoalveolar lavage. An abnormal BAL (alveolitis) was considered by the presence of ≥ 3% polymorphonuclear cells and/or ≥ 2% eosinophils.
Clinical and serologic variables recorded included: demographics, scleroderma subtype, presence of anti-topoisomerase (anti-Scl-70) and anti-centromere antibodies (ACA), duration of CYC treatment, date of death or lung transplantation, last clinic visit date, and pulmonary function test (PFT) results. Patients were classified into either limited cutaneous scleroderma (lcSSc), if skin changes were limited to the distal limbs and/or face, or diffuse cutaneous scleroderma (dcSSc) if skin changes were on the proximal limbs and/or trunk [9, 10]. Pulmonary arterial hypertension was defined as either an estimated right ventricular systolic pressures of ≥ 40mmHg by Doppler echocardiography (ECHO) or a resting mean pulmonary arterial pressure (PAP) of > 25 mmHg with an associated pulmonary capillary wedge pressure < 15 mmHg by cardiac catheterization. Age at scleroderma onset was calculated from date of birth until the date of first non-Raynaud’s symptom. Disease duration was defined as duration from the first non-Raynaud’s symptom until ILD diagnosis. The date used for ILD diagnosis was the date of the PFT closest to initiation of CYC therapy. Follow-up period was defined as the date from CYC initiation until the date meeting one of the three defined endpoints (death, lung transplantation, or lung function decline). For those who did not have an event, the follow-up period was calculated from the date of CYC initiation until the latest available follow-up date (either last clinic visit or last PFT date, whichever occurred later).
Ethics Statement
Clinical investigation of the scleroderma registry was with approval by Johns Hopkins institutional review board and was in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants involved in this study.
CYC Treatment
The dose and duration of CYC treatment was determined by the individual treating physicians. It has been the typical practice of the authors to initiate 1mg/kg of body weight of daily oral CYC and to titrate to a goal of 2mg/kg of body weight provided no adverse events, toxicity, or signs of intolerance occurred. The typical recommended duration of CYC treatment increased over the study period from 6 to 18 months, reflecting the authors (FW, RW, LH) change in clinical practice. Duration of CYC therapy was determined by record review.
Pulmonary Function Tests (PFTs)
PFTs were routinely followed every 4-6 months; PFTs include spirometry, measurements of lung volumes, and a single-breath carbon monoxide diffusing capacity (DLco). Forced vital capacity (FVC), total lung capacity (TLC), and DLco were obtained from medical chart review and the center’s database. Although PFTs were not all performed at the Johns Hopkins center, all PFT data was standardized to same reference values [11-13]. PFT data used in this analysis were: 1. the PFT at the time of ILD diagnosis; 2. the PFT closest to the defined endpoints, occurring at least 6 months after of CYC treatment and 3. the latest PFT, for patients not meeting any endpoint. PFT results after lung transplantation were excluded from analysis.
Endpoints
We defined 3 endpoints of interest: significant lung function decline, lung transplantation, and death. The endpoints had to occur at least 6 months after CYC initiation. To assess for possible survival bias, we compared the demographic features our study population to those with a positive BAL but with less than 2 years of follow up data. A clinically significant decline in lung function was defined as a decline of ≥ 10% predicted FVC or DLco on follow-up PFT compared to PFT values at ILD diagnosis; a clinically significant improvement was defined as an improvement of ≥ 10% predicted FVC or DLco on follow-up PFT compared to PFT [6]. Time until lung function decline, lung transplantation, or death was calculated from the date of CYC initiation until the date an endpoint was met. The last follow-up date for patients who did not meet a defined endpoint was either as the date of their last clinic visit or the date of last follow-up PFT, whichever date occurred later.
Statistical Analysis
Continuous data are presented as a mean ± standard deviation (SD), or as median (range), for non-normally distributed data unless otherwise specified. Fisher’s exact test and unpaired t-test were used to analyze the two groups with respect to age of onset, gender, race and mortality. Paired t-test was used to analyze change in percent predicted FVC and DLco from baseline and follow-up. Kaplan Meier graphs were generated for individual and combined endpoints.
RESULTS
Ninety three patients seen at the Johns Hopkins Scleroderma Center from March 1991 to April 2006 were identified as having received CYC for ILD. Among those screened, 38 patients met our pre-review criteria. Reasons for exclusion included: a treatment period of less than 6 months (24 patients), a follow-up period of less than 2 years (20 patients), and incomplete data (11 patients). Of the 20 patients with less than 2 years of follow-up, 6 had died compared with 5 out of 38 patients who died or had a lung transplant (p=0.16). No significant differences were seen by age at SSc onset, gender, or race between the study population and the 20 patients with less than 2 years of follow-up. The six deaths that occurred in this comparison group included 2 due to severe ILD complicated by severe cardiomyopathy, 1 due to a failed lung transplant for severe ILD, and 1 from a combination of severe ILD and PAH. One patient died of a suicide and one died of likely pulmonary embolus.
The baseline characteristics of the 38 patients are presented in Table 1. All patients met ACR criteria for the diagnosis of scleroderma [9]. The majority were women (26 patients, 68%) and Caucasians (28 patients, 74%) with an age of onset of 44.3 ± 11.4 years. Twenty-two dcSSc and 16 lcSSc were followed for an average of 5.1 years (range 2.3 -10.8 years) from start of CYC until end of study follow-up period. Eighteen out of 36 patients (50%) screened had a positive anti-topoisomerase antibody and only one out of 35 patients screened was anit-centromere positive. The median duration of CYC treatment was 1.4 years (range 6 months-6.7 years). Twelve patients had up to a year of CYC, 17 had 1-2 years, 9 had >2 years of treatment. Among the 9 patients treated for >2 years; 1 had 26 months, 2 had 30 months, 1 had 32 months, 1 had 35 months, 1 had 38 months, 2 had 41 months, and 1 had 80 months of CYC. Thirty-seven out of 38 patients had abnormal baseline PFTs; one patient with a normal baseline PFT had an abnormal BAL.
Baseline Clinical Characteristics of Study Population (N=38)
| Characteristics | Results |
|---|---|
| Age of SSc onset mean ± SD (range), in years | 44.3 ± 11.4 (21-74) |
| Disease duration at ILD diagnosis mean ± SD (range), in years | 4.6 ± 4.6 (0-23) |
| Gender | |
| Female: Male, n (%) | 26 (68): 12 (32) |
| Race | |
| Caucasian, n (%) | 28 (74) |
| African American, n (%) | 7(18) |
| Other, n (%) | 3 (8) |
| Subtype | |
| lcSSc: dcSSc, n (%) | 16 (42): 22 (58) |
| Autoantibody Status | |
| positive ACA, n | 1 |
| positive anti-TPO, n | 18 |
| Baseline % predicted FVC, mean ± SD (range) | 63.3 ± 12.4 (39.4-95.5) |
| Baseline % predicted DLco, mean ± SD (range) | 52.6 ± 16.4 (26.2-102.1) |
| Follow-up period mean ± SD (range), in years | 5.1 ± 2.1 (2.3-10.8) |
| Duration of CYC therapy mean ± SD (range), in months | 20.3 ± 13.6 (6 months-6.7 years) |
Legend: ACA- anti-centromere antibody: 3 missing ; Anti-TPO-anti-topoisomerase antibody: 2 missing; SSc- systemicscleroderma; CYC-cyclophosphamide; FVC- forced vital capacity; DLco-single-breath carbon monoxide diffusing capacity; lcSSc- limited cutaneous scleroderma; dcSSc-diffuse cutaneous scleroderma.
PFT Endpoint
Forced Vital Capacity
The mean % predicted FVC at follow-up was 62.3 ± 19.6%, which was a mean decline of 1.0% (95% CI of -6.2 to 4.2%) from baseline. There was no significant difference between the % predicted FVC at baseline and at follow-up (p=NS). Nine (24%) patients had a significant improvement, 12 (31%) had a significant decline, and 17 (45%) had stable lung function at follow-up.
At a median of 4.1 years from start of CYC therapy, sixty-eight percent of patients had a stable FVC while 12 out of 38 patients (32%) had a significant decline (Fig. 1). Among the 12 patients with a significant decline in FVC, 10 were treated for greater than 1 year with CYC and 4 had greater than 2 years of CYC treatment.
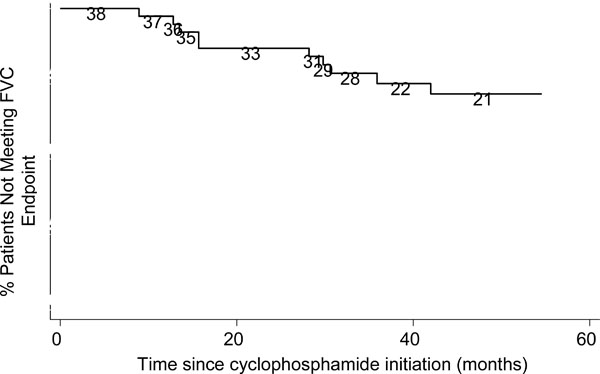
Kaplan-Meier Graph for % Predicted FVC decline. Numbers are persons at risk at each interval.
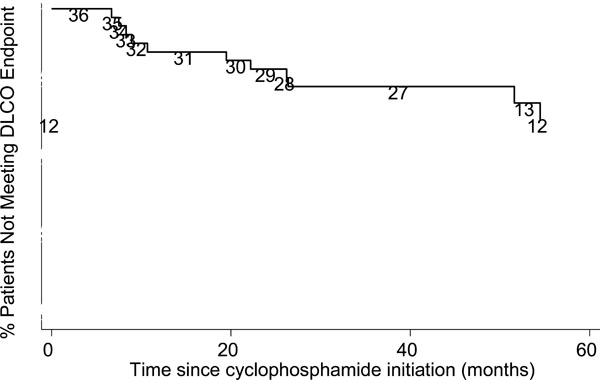
Kaplan-Meier Graph for % Predicted DLco decline. Numbers are persons at risk at each interval.
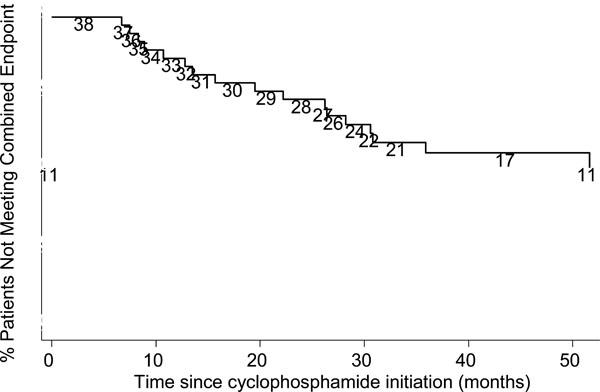
Kaplan-Meier Graph of combined endpoint. Numbers are persons at risk at each interval.
Single Breath Carbon Monoxide Diffusing Capacity
Values for DLco were not available on follow-up PFT for 2 patients. The % predicted DLco at follow-up was 54.8 ± 26.4%, which declined 1.5 % from baseline (95% CI of [-9.8% to 6.7%], p=NS). Sixty-seven percent (24/36) of the study population had stable DLco. Thirty three percent of patients (12/36) experienced a significant DLco decline in a median of 3.9 years (range 7 months - 8.4 years) (Fig. 2). Five out of these 12 patients had a diagnosis of pulmonary hypertension by ECHO and 2 patients had their diagnosis subsequently confirmed by cardiac catheterization; 9 out of 12 patients had greater than 1 year of CYC treatment and 4 had greater than 2 years of treatment.
Long Term Survival and Lung Transplantation
Three subjects died during the study period. Two patients died as a result of pulmonary arterial hypertension. Both patients experienced a significant drop in DLco at a follow up period of 7.5 months, with a DLco drop of 37.4%, and 26.4 months, with a DLco drop of 17.7%; one patient also had a significant drop in FVC at 3.5 years of 13.9 %. One patient died of an unknown cause at a follow-up of greater than 5 years and had experienced a significant drop in DLco 7 months into treatment; the % predicted FVC and DLco on the last available PFT was 110.5% and 78% respectively. This patient had no evidence of PAH at last ECHO.
Two patients received bilateral lung transplants for severe restrictive lung disease. One patient had a severe restrictive lung disease 32 months from initiation of CYC; the % predicted FVC at time of transplant was 44.3% compared with their baseline of 43%. There was no DLco data available for this patient at time of transplantation. Another patient, transplanted 48 months after initiation of CYC, had a significant decline only in FVC (drop from 71.1% to 27.1% at 36 months) with a stable DLco (49.5% at baseline and 44.7% at follow up).
Combined Lung Function, Survival, and Lung Transplantation Endpoints
Seventeen out of 38 patients (45%) experienced lung function decline, death, or a lung transplant at a median of 2.95 years (range 6.7 months – 8.4 years) from CYC initiation (Fig. 3). While lung function decline occurred more frequently by 2 years, lung transplantation or death occurred with greater frequency after 2 years.
DISCUSSION
We found that 55 and 75% of CYC-treated SSc patients with active ILD have a stable FVC and DLco at a median follow-up of 4.5 years, respectively. We also found that the 6-year survival rate post-CYC treatment is 60%, however, around 40% of SSc patients with ILD have continued lung function decline despite treatment.
The median survival of SSc-ILD, defined as fibrosis on a chest X ray and a % predicted FVC<55%, is estimated at 3 years from the onset of organ involvement. CYC treatment is thought to be effective at limiting lung function decline in SSc patients with active ILD. The first randomized, controlled trial of SSc patients with active ILD showed a significant, although modest improvement in lung function after CYC treatment compared with placebo at one year, but not at 2 years of follow-up [3, 4]. Approximately 7% of CYC-treated patients had a clinically significant decline in lung function, defined as ≥10% change in percent predicted FVC from baseline to 1 year; the percentage of CYC-treated patients with a clinically significant decline in lung function at 2 years was not reported. We found that 3% and 14% of treated patients had a ≥10% percent predicted FVC decline at 1 and 2 years follow-up, respectively; unfortunately, there was no placebo group to provide a comparison. However, data from the placebo arm of a randomized clinical trial of SSc-ILD revealed an average rate of decline in % predicted FVC of –2.01 per unit score for fibrosis [3].
There are few studies of CYC-treated SSc patients with ILD having lung function follow-up of greater than 2 years [14-16]. A retrospective study by White et al. of SSc patients with ILD seen at our Center from 1991-1998 showed that CYC-treated patients followed for a median of 16 months had stabilization in lung function, as measured by FVC and DLco [6]. Another study of 13 SSc ILD patients, who were treated with intravenous (IV) monthly pulse CYC and methylprednisolone, showed that 60% had stabilization in % predicted FVC or DLco at 48 months [15]. Other studies of 13 and 14 SSc ILD patients also yielded similar results at 12 and 26 months follow-up, respectively [14, 16]. We observed a large group of SSc patients who had serial PFTs with a mean follow-up of 4 years and found around 60% of patients, a similar proportion to these other studies, had long term lung function stabilization. Our study also extends the work by White et al. and includes a broader, more clinically applicable definition of ILD as opposed to defining ILD only as patients who have had an abnormal BAL or lung biopsy result. Our study population also included patients with milder restrictive lung disease who were treated with a longer period of CYC compared with the population evaluated by White et al.
Few studies have examined the effect of CYC on survival. In the Scleroderma Lung Study, the CYC-treated patients had a significant improvement in combined endpoint of death and FVC at 12 months compared with placebo and 57 out of 73 CYC-treated patients (78%) were alive at 2 years [3, 4]. In a retrospective study of SSc patients by White et al., 39 CYC-treated patients identified as having ILD by a BAL had a median survival rate of 89%; 10 patients survived for up to 4.8 years [6]. However, these patients had variable severity of ILD and pulmonary hypertension.
We report in this study our experience from a large number of well-characterized CYC-treated patients followed longer than two years with respect to lung function and mortality. Limitations in our study include its retrospective design and analysis of a selected group of SSc patients who were treated with CYC and who survived for two or more years. Thus, while this study population does not reflect those who have early mortality, it does give insight into the projected long-term course of patients who do complete a course of CYC treatment and who survive for longer than two years. We could not find variables that predicted adverse outcomes, but the study population was not large enough to reliably evaluate subgroups. Although there seemed to be no significant differences in mortality between our study population compared to the group of patients with < 2 years of follow-up, there were more deaths attributable to ILD in this latter group, suggesting that ILD was less severe in our study population. Moreover, 2 out of the 3 deaths that occurred in our study population were due to PAH. This may indicate that patients who survive beyond 2 years are less likely to die from ILD.
Our findings imply that CYC limits the progression of lung disease in many, but not all SSc patients. Roughly one third of patients experience continued decline despite “standard of care treatment”, highlighting the need for continued lung function monitoring and alternative treatments. It will be important to determine if other anti-fibrotic, anti-inflammatory, or immunosuppressive agents will be superior or safer than CYC and if maintenance therapy is required after inducing a clinical response with CYC therapy. This study also did not address the strategy of maintenance immunosuppressive therapy after a course of CYC or re-treatment with another agent in the setting of evidence of progressive disease despite CYC therapy. We found that SSc patients treated with CYC for periods greater than one year did not have significantly different lung function outcomes compared to those treated less than 12 months, albeit the size of our study population was small. We cannot exclude that a proportion of patients who experienced a decline in DLco had unrecognized pulmonary arterial hypertension.
In conclusion, we report our experience with CYC-treated SSc patients with ILD and find that most patients have a good long-term prognosis, both in terms of lung function and survival. However, there remain a proportion of patients who will have lung function decline despite treatment with CYC.

