All published articles of this journal are available on ScienceDirect.

Systemic Administration of Tolerogenic Dendritic Cells Ameliorates Murine Inflammatory Arthritis
Abstract
The expression of various cell surface molecules and the production of certain cytokines are important mechanisms by which dendritic cells (DC) are able to bias immune responses. This paper describes the effects of the inflammatory cytokine tumor necrosis factor (TNF)-αon DC phenotype and function. TNF-αtreatment resulted in upregulation of MHC class II and CD86 in the absence of increased cell surface CD40 and CD80 or the production of IL-12. Additionally TNF-αtreated cells were able to bias T cell responses towards an anti-inflammatory profile. On a note of caution this tolerogenic phenotype of the DC was not stable upon subsequent TLR-4 ligation as a 4 hour pulse of the TNF-α treated DC with lipopolysaccharide (LPS) resulted in the restoration of IL-12 production and an enhancement of their T cell stimulatory capacity which resulted in an increased IFN-γproduction. However, TNF-αtreated DC, when administered in vivo, were shown to ameliorate disease in collagen induced arthritis, an experimental model of inflammatory joint disease. Mice receiving TNF-αtreated DC but not LPS matured DC had a delayed onset, and significantly reduced severity, of arthritis. Disease suppression was associated with reduced levels of collagen specific IgG2a and decreased inflammatory cell infiltration into affected joints. In summary the treatment of DC with TNF-αgenerates an antigen presenting cell with a phenotype that can reduce the pro-inflammatory response and direct the immune system towards a disease modifying, anti-inflammatory state.
INTRODUCTION
Dendritic cells are powerful antigen presenting cells (APC) that are adept at both stimulating naïve T cells and controlling the quality of subsequent immune responses. This control is determined by the subset of DC, and the type and duration of signals they receive. In the absence of external stimulation, DC reside in peripheral tissues in an immature state and express relatively low levels of MHC class II and no co-stimulatorymolecules[1]. These immature DC are essential mediators of peripheral tolerance to self and other non-hazardous foreign antigens [2]. The general view is that tolerance results from the presentation of antigens in the absence of full co-stimulation requirements to drive T cell activation. In response to infection, DC undergoes both phenotypic and functional changes. The antigen presenting capacities of the DC are enhanced via the up-regulation of cell surface expression of co-stimulatory molecules (CD80, CD86 and CD40) and other molecules that promote DC-T cell clustering (CD54 and CD58). Activated DC also alter their profile of chemokine receptors to facilitate homing to lymphoid organs, and secrete various pro-inflammatory cytokines [3]. Many factors present as a result of tissue damage, inflammation and infection are known to influence DC maturation. Such factors differ in the signals they provide to the DC therefore controlling their effector functions and hence the outcome of the immune response [4]. Thus, modulation of DC activation provides a potential therapeutic strategy to limit unwanted inflammatory responses.
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by joint inflammation and progressive cartilage and bone erosion [5]. Inflammatory cytokines, such as TNF-α and IL-1 are vital mediators in the pathogenesis of RA, secreted predominantly by synovial macrophages and fibroblasts. In addition, increased levels of Th1 cytokines are involved[6, 7].This knowledge of the mechanisms of pathogenesis led to the fundamental breakthrough of the use of anti-TNF-α reagents as efficient therapy for RA [8-1]. However, this requires repeated administration thus incurring substantial costs to health care provision. Alternatively, the promotion or enhancement of an anti-inflammatory response is considered to contribute to a successful therapeutic strategy [12-14].
Collagen induced arthritis (CIA) is an experimental animal model of RA characterized by inflammatory cell infiltration and synovial hyperplasia leading to progressive bone and cartilage degradation [15]. In susceptible mouse strains inflammatory joint disease is mediated by a pro-inflammatory response directed towards type II collagen (CII) [16-18] in concert with the production of CII-specific complement fixing antibodies [19, 20]. In light of the potential of DC to bias immune responses, we have studied the possibility of targeting these cells to modulate T cell responses in experimental arthritis. Our results presented in this paper build on the concept that TNF-α modulation is efficacious, but innovatively we propose that short term TNF-α administration to DC can be equally effective.
MATERIALS AND METHODOLOGY
Animals
Specific pathogen free male DBA-1 mice aged 8-12 weeks were obtained from Harlan Olac, Bicester, UK and were conventionally housed and fed ad libitum. All experiments were performed according to UK Home Office guidelines.
Generation and Culture of Bone Marrow Derived Dendritic Cells
Bone marrow (BM) derived DC were generated as previously described [21]. Briefly, BM cells were harvested from the femura and tibia of both hind legs and cultured in RPMI 1640 (Gibco, Paisley, UK) containing 10% FCS (Harlan Sera-Lab, Leicestershire, UK), 1% HEPES buffer, 4mM L-glutamine (GIBCO-BRL, Life Technologies, Paisley, UK) 100 IU/ml benzyl penicillin (Sigma Chemical Co., Poole, UK), 100μg/ml streptomycin sulphate (Sigma), and 20ng/ml of GM-CSF (Peprotech, London, UK). After 7 days DC were harvested and left untreated or matured with 1μg/ml of lipopolysaccharide (LPS) (E. coli derived; Sigma) or 500U of TNF-α (Peprotech)for 4h. Where indicated, DC were pulsed for 4h with 40μg/ml of OVA (Sigma), 1μg/ml LPS or a synthetic peptide containing the immunodominant epitope of type II collagen (CIIp; corresponding to amino acids 259-270; Alta Bioscience, Birmingham, UK). DC were washed extensively, and either phenotyped by flow cytometry and their cytokine production assessed, or were injected into mice.
Flow Cytometry
Unstimulated, LPS matured or TNF-α treated DC were defined by phenotypic analysis for expression of CD11c, CD40, CD80, CD86 and MHCII. In addition, the production of IL-12 by DC was assessed by intracellular staining. Briefly, cells were incubated with 2μM of Golgi-stop (Sigma) for 4h, washed, re-suspended in staining buffer (Phosphate buffered saline (PBS) containing 2% foetal calf serum (FCS) and 2% bovine serum albumin), and incubated with anti-CD11c for 30 minutes on ice. Cells were fixed in fixation buffer (PBS containing 4% paraformaldehyde (PFA); pH 7.4-7.6) for 20 minutes at room temperature and washed twice in permeabilization buffer (PBS containing 2% FCS and 0.5% saponin). Cells were re-suspended in staining buffer and incubated with phycoerythrin (PE)- conjugated anti-mouse IL-12/IL-23 (p40) for 30 minutes at room temperature prior to washing and analysis. All antibodies were directly labeled and obtained from BD PharMingen (BD PharMingen, San Diego, CA) and used at dilution of 1:200 except anti-IL-12 which was used at 1:100. Cells were analyzed by flow cytometry using Cell Quest software (FACScan; BD PharMingen). In the cultures 75-80% of the cells stained positive for CD11c.
Injection of Antigen Loaded DC
Groups of mice were injected subcutaneously (sc) at the base of tail with 1 x 106 DC that had been LPS or TNF treated and pulsed with ovalbumin (ova) or CIIp. Control mice received injections of PBS. Ten days later, spleen and draining lymph nodes were removed, single cell suspensions prepared in α-MEM (Cambrex Bioscience) supplemented with 4mM L-glutamine (GIBCO-BRL), 100U/ml benzyl penicillin (Sigma), 100μg/ml streptomycin sulphate (Sigma), 5 x 10-5 M 2-mercaptoethanol (Sigma) and 20 mM HEPES buffer (Sigma) and 0.5% fresh normal mouse serum (NMS). Cells were incubated at 37(C in a humidified atmosphere of 5% CO2 in 1ml cultures in 48 well plates (ICN Biomedicals) at a concentration of 2.5 x 106 cells/ml, and stimulated with 80μg/ml of OVA or CIIp. Three days later cell proliferation and cytokine production were analysed.
Proliferation Assays
For the analysis of proliferation by spleen and lymph node cells, 100μl samples from each culture were transferred to a 96 well round bottomed plate (Nunclon, Denmark) and pulsed for 16h with 0.5μCi/ml of (3H(-thymidine (specific activity 25Ci/mMol; Amersham International Ltd, Amersham, UK). Cells were harvested onto glass filter mats (Wallac, Turku, Finland) using a Mach III Harvester 96 (Tomtec. Orange, USA), and the incorporation of 3H-thymidine was measured in counts per minute (cpm) using a liquid scintillation counter (Microbeta plus: Wallac).
Cytokine Production
Cytokine production by cultured spleen and lymph node cells was measured by a previously described cell based ELISA protocol (CelELISA) [22]. Briefly, 100μl samples were harvested from cultures on the indicated day, and incubated for an additional 24h in 96 well plates (Maxisorb Immunoplates, Nunc Ltd, Rosklide, Denmark) previously coated overnight at 4(C with monoclonal anti-cytokine Ab. After washing, bound cytokines were detected by the addition of biotinylated anti-cytokine monoclonal Ab directed to non-overlapping epitopes. Coating Ab were rat anti-mouse IL-4 (Clone 11B11), IL-5 (Clone TRFK5), IFN-γ (Clone R4-6A2) and IL-10 (Clone JES5-SXCI). Detection Ab were IL-4 (Clone BVD6-24G2) IL-5 (Clone TRFK4), IFN-γ (Clone XMG1.2) and IL-10 (Clone JES5-2A5). Streptavidin-horse radish peroxidase (HRP) reagent (Sigma) and 3,3,5,5’-tetramethylbenzidine were used as the enzyme and substrate respectively. The reaction was stopped with 2M H2SO4 and the plates read at 450nm. The levels of each cytokine in the samples were determined by regression analysis from a standard curve constructed using recombinant cytokine (IL-4, IL-5, IFN-γ, IL-10). All Ab/recombinant cytokines were from PharMingen, BD except IL-10 (Biosource International, USA).
Measurement of Type II Collagen Antibody Responses
Serum levels of anti CII Ab were measured by standard ELISA. Briefly, 96 well high binding plates (Greinerbio-one, UK) were coated with murine CII (20μg/ml) for 1h at 37(C and blocked with 10% FCS for 1h at room temperature. Sera were diluted to 1/10,000 and incubated at 37(C for 2h. After washing, bound Ab was detected by incubating the plates with HRP-conjugated anti-mouse IgG or anti-mouse IgG isotype Ab (IgG1 or IgG2a; all from Serotec, UK). Thereafter plates were washed, incubated with 100μl/well of O-phenylenediamine dihydrochloride (Sigma) diluted in citric-phosphate buffer, the reaction stopped by adding 50μl/well of 2M sulphuric acid and the plates read at 492nm. Antibody units for murine CII were determined using a reference serum created from pooled sera of arthritic mice. This serum was assigned an arbitrary level of 100 units of antigen specific Ab.
Induction of Arthritis
Type II collagen was prepared from bovine nasal septum as previously described [23]. CII was dissolved in 0.1M acetic acid at 4°C at a concentration of 4mg/ml. DBA-1 mice were injected intradermally (id) at the base of the tail with 100μl of CII emulsified in an equal volume of complete Freunds adjuvant (CFA; Difco, UK). On day 21, mice received a booster injection id at the base of the tail with 100μl of collagen in incomplete Freunds adjuvant (Sigma). Mice were scored for clinical signs of arthritis in the limb joints by macroscopic examination twice weekly. Arthritis severity in each paw was graded according to the established scoring system [24, 25], 0 = no arthritis, 1 = 1 inflamed digit, 2 = 2 inflamed digits and/or erythema and mild swelling of the footpad, 3 = > 2 digits and footpad inflamed, 4 = all digits and footpad inflamed. An arthritis score for each mouse was calculated by summing the scores for each paw. Scoring was conducted blind by an independent investigator.
Therapy of CIA with DC
Groups of 8 mice were injected sc at the base of the tail with 1 x 106 untreated; LPS or TNF-α treated DC 3 days before immunization with CII in CFA and again 3 days before the booster immunisation. Control mice received injections of PBS at these 2 time points. Mice were scored for clinical signs of disease until day 40.
Histology
Hind paws were collected and fixed in 4% PFA and decalcified in 15% EDTA and 30% glycerol. Tissue was then dehydrated in a gradient of alcohols, paraffin embedded sectioned at 5μm, mounted on glass slides and stained with hematoxylin and eosin.
Statistical Analysis
Results were compared using the Mann Whitney test. P values of < 0.05 were considered significant.
RESULTS
Phenotype of, and Cytokine Production by, TNF- α Treated DC
Initial experiments investigated the response of DC to incubation with the inflammatory cytokine TNF-α or LPS. DC cultured from BM precursor cells in GM-CSF for 7 days and treated for 4 hours with LPS or TNF-α were assessed for their cell surface phenotype and ability to produce IL-10 and IL-12. A large proportion of the BM cells were positive for CD11c (> 75%) indicating that these cells were of a DC phenotype (data not shown). As illustrated in Fig. (1), un-stimulated DC expressed MHC class II, CD80 and CD86 but limited CD40.Compared with unstimulated cells, LPS treated DC displayed elevated levels of CD80 and CD86, and a marginal increase in MHC Class II and CD40. In contrast, TNF-α treated DC displayed greatly elevated levels of MHC class II and CD86, a marginal increase in CD80 but no up-regulation of CD40. Additionally, cytokine production by the differentially stimulated DC also varied. In response to LPS stimulation DC produced enhanced levels of IL-10 and an increase in the percentage of cells expressing IL-12. However, upon treatment with TNF-α the quantity of IL-10 produced and the expression of IL-12 was the same as that produced by unstimulated DC (Fig. 2). In summary, the major differences between TNF-α treated DC and LPS activated DC are greatly elevated MHCII expression and reduced CD40 and CD80 expression concomitant with failure to secrete IL-12. Together, these findings indicate that the signals delivered through TNF-α or LPS differ in their ability to stimulate up-regulation of surface molecules and cytokine production by DC.
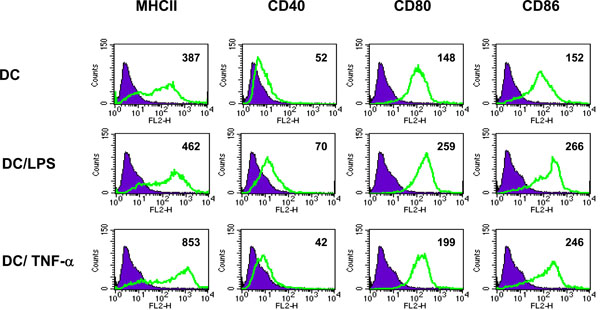
Phenotype of TNF-α treated DC. DC were unstimulated, activated with LPS (1µg/ml for 4h) or treated with TNF-α (500U/ml for 4h) and analyzed by flow cytometry for cell surface expression of MHC class II, CD40, CD80 and CD86. Filled histograms represent isotype matched control antibody staining. Numbers indicate MFI of stained cells. Data shown is representative of 2 separate experiments.
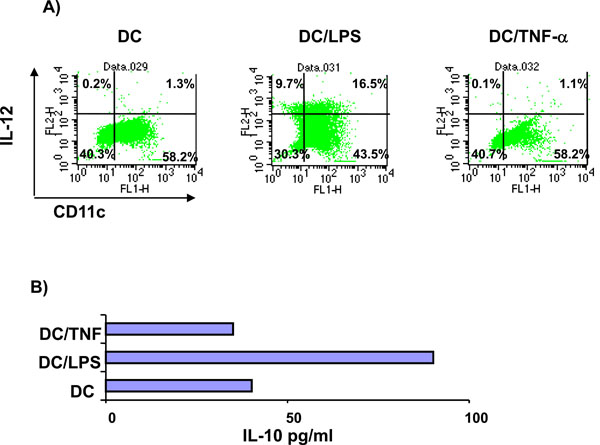
Cytokine production by TNF-α treated DC. Unstimulated, LPS activated (1µg/ml for 4h) or TNF-α treated DC (500U/ml for 4h) were analyzed for the production of IL-12 and IL-10. (A) DC were stained for cell surface expression of CD11c and intracellular IL-12 and analyzed by flow cytometry. Quadrants were set using isotype control staining. Values indicate the percentage of cells in each quadrant. (B) Supernatants from the unstimulated, LPS activated or TNF-α treated DC were tested for IL-10 production by ELISA. Data shown are mean IL-10 production from duplicate samples and are representative of 2 separate experiments.
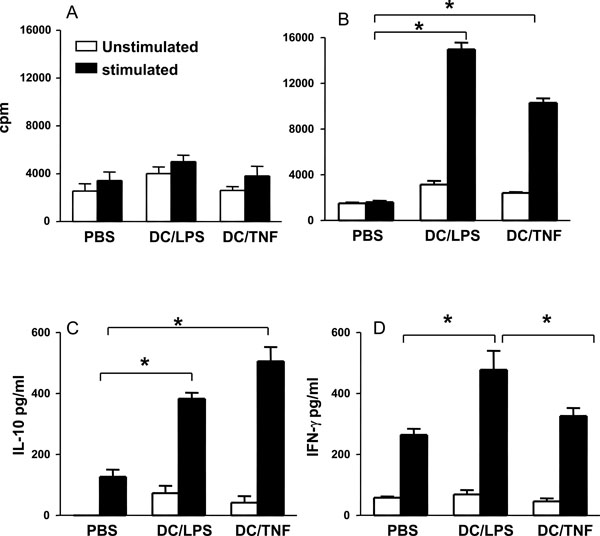
TNF-α treated DC bias T cells towards an anti-inflammatory phenotype. Groups of mice were injected sc at the base of tail with 1 x 106 DC that had been LPS activated or TNF treated and pulsed with CIIp (A) or Ova (B). Control mice received injections of PBS.Ten days later, spleen and draining lymph nodes were removed. Cell cultures were prepared and stimulated with CIIp (A) or ova (80µg/ml) (B, C, D). After 4 days proliferation was assessed and cytokine production (IFN-γ, and IL-10) measured. Bars represent the mean ± SEM (* = p < 0.05 by Mann Whitney test).
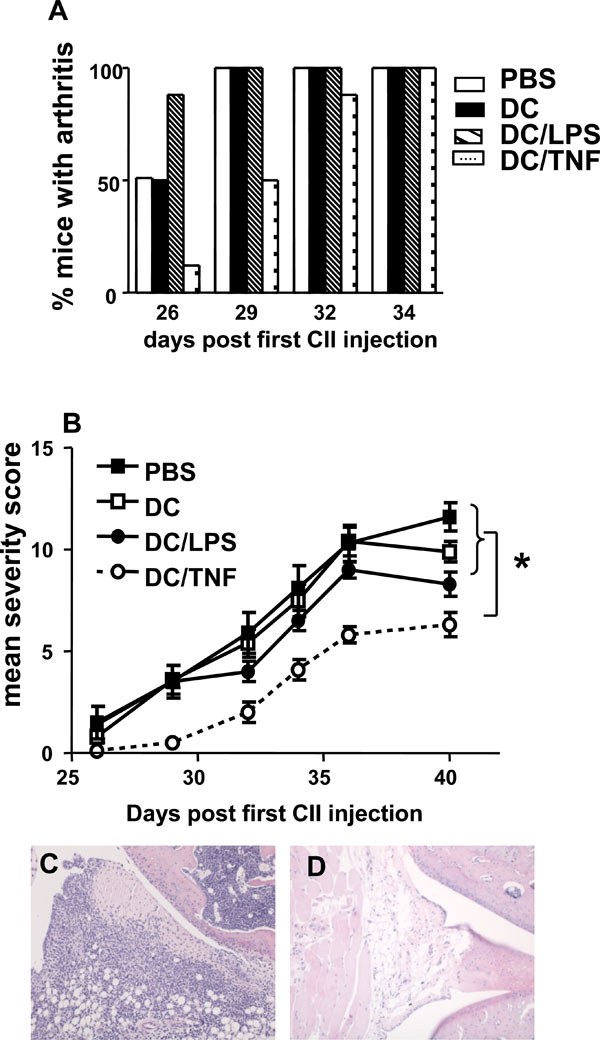
Immunotherapy of murine inflammatory arthritis. Groups of 8 mice were injected sc at the base of tail with 1 x 106 untreated,LPS activated or TNF-α treated DC 3 days prior to immunization with CII in CFA and again 3 days before the booster immunisation.Control mice received injections of PBS. Mice were scored for clinical signs of disease until day 40. (A) The % of mice with arthritis in each group over time. (B) The mean arthritis severity score in each group over time. * = p < 0.05 by Mann Whitney test. (C) Transverse section through stifle joint of PBS treated mouse stained with H&E demonstrating inflammatory cell infiltration. (D) Transverse section through stifle joint of TNF-αtreated DC injected mouse indicating much reduced cellular infiltration. Magnification x10.
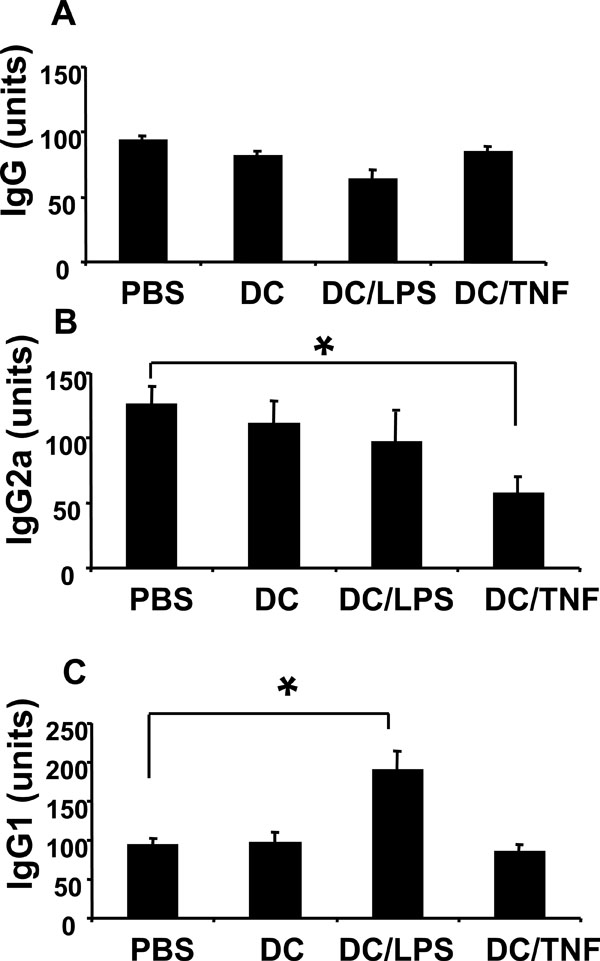
Reduced CII specific IgG2a in sera obtained from mice injected with TNF-αtreated DC. Sera were collected at termination of the disease study, and levels of CII specific antibody determined by ELISA. (A) Total IgG. (B) IgG2a isotype. (C) IgG1 isotype. The antibody units reflect the relative amounts of CII specific antibody compared to a standard control serum (pooled sera from arthritic mice). This standard represented 100 units of antigen specific antibody. Levels of IgG2a and IgG1 have been normalised against amounts of total IgG detected in each sample. Values are the mean (n=8/group) ± SEM (* = p < 0.05 by Mann Whitney test).
TNF-α Treated DC Bias T Cells Towards an Anti-Inflammatory Phenotype
In order to investigate the antigen presenting function of each of the DC types in vivo, mice were injected with LPS activated or TNF-α treated DC that had been pulsed with the immunodominant epitope corresponding to amino acids 259-270 of collagen (CIIp) or OVA. Ten days after immunization, cells from spleen and lymph nodes were assessed for their responses to restimulation with OVA or CIIp.Cells from mice that had received CIIp pulsed DC, regardless of their pretreatment, failed to respond to CIIp, suggesting that BM-DC are poor antigen presenting cells for CIIp in vivo (Fig. 3A) confirming the in vitro observations by Holmdahl and colleagues [26]. However, spleen and lymph node cells were capable of vigorous proliferation to the T cell mitogen Con A (data not shown) thus demonstrating their viability. By contrast, both LPS activated and TNF-α treated DC were clearly capable of presenting OVA in vivo ascells from mice that were injected with OVA pulsed DC proliferated in response to antigen in vitro (Fig. 3B). The quality of the immune response generated by the differentially treated DC was assessed by measuring the levels of cytokines produced by spleen and lymph node cells stimulated with OVA in vitro. The production of the Th2- associated cytokine IL-4 was below the detection limit of the assay for all groups tested. However, as illustrated in Fig. (3C) cells from mice that had received OVA pulsed TNF-α treated DC produced higher levels of IL-10 as compared to cells derived from mice receiving OVA pulsed LPS matured DC although this difference did not reach statistical significance. However, cells from mice receiving antigen pulsed TNF-α treated DC produced significantly less IFN-γ than cells derived from mice injected with LPS matured DC (Fig. 3D). These results indicate that TNF-α treated DC bias the resulting immune response towards an anti-inflammatory phenotype as characterized by significantly lower IFN-γ production and elevated levels of IL-10.
In a separate experiment, TNF-α treated DC were pulsed with LPS for an additional 4h and were subsequently assessed for their ability to secrete IL-12 (by intracellular cytokine staining) and were also used to stimulate T cells. This pulsing of TNF-α treated DC with LPS was carried out to determine whether their ability to enhance anti-inflammatory cytokine production could be overridden by other potent inflammatory stimuli (danger signals). Interestingly, the production of IL-12 varied markedly between differentially stimulated DC. As observed previously the % of IL-12 producing DC in cultures that were stimulated with TNF-α (5%), was again comparable to the low level observed in untreated cells (4%;Table 1). By contrast to these baseline levels the proportion of IL-12 producing cells was again increased in cultures of DC that were matured with LPS (13%). Strikingly, the subsequent incubation of TNF-α treated DC with additional LPS resulted in a marked increase in the percent of IL-12 producing cells (68%). In addition, when these DC were used to stimulate T cells the response observed was increased proliferation and enhanced IFN-γ production compared to T cells stimulated with TNF-α treated DC that were not exposed to additional LPS (Table 1). These findings demonstrate that LPS is able to stimulate TNF-α treated DC to produce IL-12 and that the anti-inflammatory cytokine enhancing properties of TNF-α treated DC is lost.
Summary of DC Phenotype and Resulting T Cell Stimulatory Capacity
| DC Treatment | % CD11c+, IL-12+ | T Cell Stimulation (CPM ± SEM) | IFN-γ Production (pg/ml ± SEM) |
|---|---|---|---|
| Untreated | 4 | 3100 ± 245 | 320 ± 126 |
| LPS matured | 13 | 21050 ± 230 | 3840 ± 355 |
| TNF-α | 5 | 6465 ± 322 | 1255 ± 585 |
| TNF-α +LPS | 68 | 22455 ± 365 | 6215 ± 245 |
DC were untreated, matured with LPS (1µg/ml for 4h) or treated with TNF-α (500U/ml for 4h). In addition, TNFα treated DC were pulsed for a further 4h with LPS (1µg/ml). DC were stained for cell surface expression of CD11c and intracellular IL-12 and analyzed by flow cytometry. Values in the first column represent the % of CD11c+ and IL-12+ cells.Data from a representative experiment (of 2 performed) are shown. Ova primed T-cells (>90% CD3+ by flow cytometry) were stimulated for 4 days with DC treated as above. DC were loaded with ova by pulsing 1 x 106 cells with 80 μg/ml of ova for 4h. The DC:T cell ratio was 1:20 in each test. The second column represents the proliferation of cells as measured by H3-thymidine incorporation. The third column IFN γ production as measured by CelELISA. Values are mean and SEM of 3 experiments.
Immunotherapy of Murine Inflammatory Arthritis
Collagen induced arthritis is established by a pro-inflammatory response directed towards CII and the production of antigen-specific complement fixing Ab. Skewing the immune system towards a more anti-inflammatory CII response can prevent or interrupt CIA development. CIA therefore, represents an ideal model to investigate the disease modulating capacity of TNF-α treated DC bearing in mind the caveat that the stability of the DC phenotype in vivo is unknown. Previous studies have shown that DC are poor APC for CII [26-28]. We were able to confirm these observations (data not shown) and also demonstrate the inability of DC to efficiently present CIIp (Fig. 3A). In light of these findings, the capacity of unpulsed TNF-α treated DC to modulate disease in CIA was investigated. For this purpose unstimulated, LPS activated or TNF-α treated DC were injected 3 days prior to immunization with CII in CFA and again 3 days before the booster immunization. Mice that were injected with TNF-α treated DC had a delayed onset of arthritis compared to those injected with PBS, unstimulated DC or LPS activated DC (Fig. 4A). A lower incidence of arthritis in the group of mice given TNF-α treated DC occurred for up to 32 days post CII/CFA injection. Notably at day 29, considerable protection was still observed in the TNF-α treated DC group, whereas at this time point all control mice had developed arthritis. Moreover, in addition the mean severity score was significantly reduced in the TNF-α treated DC mice at day 29 and remained statistically significantly less than all other groups throughout the course of the experiment (Fig. 4B). In the mice that had received TNF-α treated DC the mean clinical score reached at the end of the experiment was 6.25 ± 0.59 compared with 11.63 ± 0.70 for mice injected with PBS. These results are supported by histology of the affected joints. Sections from PBS injected mice demonstrated pannus formation, significant cellular infiltration of soft tissue and cartilage erosion (Fig. 4C). In contrast, joints from mice receiving TNF-α treated DC appeared relatively unaffected (Fig. 4D). These results clearly demonstrate the capacity of TNF-α treated DC to modulate disease in CIA.
Reduced CII Specific IgG2a in Sera Obtained from Mice Injected with TNF-α Treated DC
Potential mechanisms responsible for disease modulation in mice injected with TNF-α treated DC were investigated. At the end of the disease study, cytokine production by and proliferative responses of spleen and lymph node cells were not statistically different between any of the treatment groups (data not shown). However, CII specific Ab production was clearly skewed in mice injected with TNF-α treated DC. Total CII specific IgG Ab titres were similar in all groups (Fig. 5A). By contrast, levels of anti CII IgG2a were significantly lower in sera from mice that received TNF-α treated DC, whilst levels of IgG1 remained at equivalent levels to control animals (Fig. 5B,5C). Together, these data indicate treatment of mice with TNF-α treated DC alters the isotype of the antigen specific Ab produced.
DISCUSSION
In order to efficiently activate naïve T cells, DC must up-regulate their expression of both MHC and co-stimulatory molecules. This is achieved following exposure to various signals provided by inflammatory cytokines and pathogen derived products [29]. Results presented here confirm that exposure to LPS results in full activation of DC, as indicated by high levels of MHC Class II, CD80 and CD86 and the production of IL-12. In contrast, DC that were treated with TNF-α failed to increase cell surface CD40 and CD80 or produce IL-12. However, CD86 was up-regulated and notably MHC class II expression was considerably higher than that observed in LPS treated DC. This suggests that the antigen presenting capacity of TNF-α treated cells is not impaired however their ability to orchestrate a pro-inflammatory T cell response may be reduced due to the lack of production of IL-12.
Our studies show that TNF-α DC were poor APC for the synthetic peptide containing the immunodominant sequence of type II collagen. This is consistent with previous studies indicating that in contrast to macrophages, which can effectively activate CII specific T cells, DC elicited poor T cell responses [26-28]. However, DC were able to process and present OVA and the resulting T cell responses differed depending on the prior treatment of the DC. Specifically TNF-α treated DC were able to bias T cells towards an anti-inflammatory phenotype, indicated by higher production of IL-10 and decreased production of IFN-γ. This cytokine profile is consistent with the observations that TNF-α treated DC do not secrete IL-12 which is essential for the development of a Th1 response. Moreover, the IL-10 measured cannot be derived from the DC themselves, as they produce less than 50 pg/ml as illustrated in Fig. (2B). Supporting the idea that TNF-α, in addition to its well established pro-inflammatory role, displays immunomodulatory properties is the recent work of Kim et al. who demonstrated that administration of exogenous TNF-α significantly downmodulated the severity of adjuvant arthritis in Lewis rats and decreased the IFN-γ secretion in response to a peptide derived from a disease-related antigen [30] Additionally, in comparison to LPS treated DC, proliferative responses induced by TNF-α treated cells were slightly decreased which may reflect their reduced expression of the co-stimulatory molecules CD80 and CD40. However, the tolerogenic phenotype and function of TNF-α treated DC is lost when such cells are incubated with LPS suggesting that the cells are not in a fully activated state and are not resistant to maturation when exposed to additional stimulation.
Based on these findings it is difficult to predict the outcome of the immune response after the administration of such cells into mice with arthritis as they may be unable to maintain their regulatory activity in the presence of ongoing inflammation, or endogenous danger signals and may indeed exacerbate the disease.
In spite of this cautionary note, the ability of these partially activated TNF-α treated DC to counteract the pathogenic response associated with experimental inflammatory arthritis was assessed. The administration of TNF-α treated DC resulted in delayed onset of arthritis and a reduced severity of disease. These results are consistent with Toes and colleagues who generated a population of TNF-α DC with a similar surface phenotype and cytokine profile [31]. Our findings differ from previous studies in which these cells were shown to suppress disease in other models of autoimmune diseases, including CIA, [31-33]in that these wereestablished only when TNF-α treated DC were loaded with the appropriate autoantigenic peptide. However, our data are consistent with the recent report that plasmid-modulated DC, not loaded with antigen, were able to suppress the development of CIA [34]. Jaen et al. also reported that LPS-matured DC could effectively prevent arthritis progression and additionally Lau et al. recently reported that LPS-activated DC could in fact give rise to CD62L-expressing regulatory T cells [34, 35]. These observations appear to be in direct contrast to our data presented here where injection of LPS-matured DC into mice led to a more rapid onset of symptoms. These contradictory results may be explained by the route or timing of DC delivery as in our studies the DC were given sc 3 days prior to disease induction, a protocol which we have shown leads to enhanced Th1 biasing, whereas in their studies Jaen et al. delivered the LPS-matured DC intraperitoneally 14 days after disease induction. This protocol has been demonstrated to induce populations of regulatory T cells which mediate the disease modifying effects [34].
In our studies, amelioration of disease was achieved by the administration of TNF-α treated DC that had not been exposed to CII-peptide as they were poor APC for this antigen in vitro. Therefore the effect of peptide pulsed DC on the course of CIA was not tested. However we postulate that these DC have the potential to acquire collagen derived peptides or other antigens in vivo that have been processed by macrophages. Alternatively, they may have the ability to enhance regulatory T cell function as previously described [36, 37]. Therefore, TNF-α treated DC may have potential for the immunotherapy of chronic autoimmune diseases where the antigens involved in the underlying pathological process are not clearly defined.
The precise way in which TNF-α treated DC are able to ameliorate disease in CIA is not completely understood. One possibility is that the differential expression of cell surface molecules may account for the efficacy of these cells. Studies suggest that failure to provide a strong CD40 signal results in T cell tolerance [38, 39] and tolerance induction is prevented by the administration of agonistic anti-CD40 mAbs [40]. As TNF-α treated DC fail to up-regulate CD40, this may result in a tolerized antigen-specific T cell response. Additionally various studies have shown an essential role for IL-12 in the induction of CIA via the development of Th1 cells. In support of this, CIA is suppressed inIL-12 knockout mice and disease susceptibility restored via the administration of exogenous IL-12 [41]. The suppression of CIA in each case was associated with reduced production of IFN-γ and CII specific IgG2a Ab. As we have demonstrated that TNF-α treated DC fail to produce IL-12 and consequently reduce antigen specific Th1 responses, this may in part explain their ability to suppress CIA.
Numerous studies have reported the amelioration of CIA by reducing the pro-inflammatory cytokines required for disease induction [18, 42, 43] or by increasing regulatory cytokines during the early phase of disease [44-46]. Indeed, anti-TNF-α treatment is reported to enhance regulatory T cell activity in patients with rheumatoid arthritis [47]. However, in our experiments, T cell proliferation and cytokine production at the end of the disease study were not significantly different between any of the treatment groups suggesting that the effect of the TNF-α treated DC is relatively short-lived. A possible explanation for this finding is that T cell responses to CII are essential in the initiation of CIA and are less involved in later stages of disease. However the reduced levels of anti CII IgG2a detected in mice that were protected from disease reflects a shift in the balance of the pro-inflammatory/anti-inflammatory cytokines mediated via the tolerogenic DC at an earlier stage in disease development.
Previous studies have shown that CII specific Ab are essential in the pathogenesis of CIA [48, 49] and in particular those of the complement fixing IgG2a isotype [19].The Ab isotypes IgG2a and IgG1 differ in their ability to engage various effector mechanisms that are essential for the induction of disease in CIA. For example, the IgG2a isotype is more effective for complement activation and the triggering of IgG Fc receptors (FcγR) than IgG1 [50]. Therefore the selective reduction in pathogenic IgG2a (as shown in this study) would be beneficial in counteracting disease development.
During the course of rheumatoid arthritis, DC present in the inflamed joint would be exposed to elevated levels of TNF-α, and in light of our results expected to acquire a tolerogenic phenotype. This is plainly not the case. However a recent study demonstrated that DC activated via toll like receptor recognition of pathogen associated molecules adopted a fully mature phenotype with the ability to produce IL-12 and prime a Th1 response [51]. Consistent with this, studies presented here indicate that partially activated TNF-α treated DC can be induced to produce IL-12 via subsequent stimulation with LPS. As there are high levels of toll like receptors expressed in the rheumatoid joint [52, 53], signaling via these molecules, or via cytokines such as IL-1 which share common signaling pathways [54], could contribute to maintaining DC in a fully mature state, thus perpetuating a pro-inflammatory response and disease progression. Moreover recent clinical data suggest that DC isolated from patients with active RA are less able to secrete TNF-α when compared to healthy subjects perhaps indicating a failure to generate DC which can control unwanted pro-inflammatory T cell responses [55]. Alternatively, short term exposure to TNF-α may generate DC capable of expanding IL-10 producing (regulatory) T cells with the capacity to feedback and maintain DC in a semi-mature state. Future work will focus on the exact mechanism, and more specifically the site of action of TNF- α treated DC. However our findings have indicated that TNF- α treated DC can modulate the course of RA, thus adding this autoimmune condition to the growing list of diseases with the potential to be controlled by DC immunotherapy [56, 57].
CONCLUSIONS
Experiments presented in this study clearly illustrate the potential of TNF-α treated DC to ameliorate CIA. Although questions remain to be answered, with regard to the phenotype of the most potent DC population, the stability of the tolerogenic DC in vivo or the precise mechanism of their action, the immunotherapeutic strategy outlined here does not rely on pulsing of DC with antigen and therefore may be appropriate for use in the therapy of autoimmune diseases for which a single autoantigen has not been identified.
ACKNOWLEDGEMENTS
The authors thank Cath Hall for excellent technical assistance and Antonio Manzo, Department of Rheumatology,Guys Hospital, London for histological analysis. LH was funded by a postgraduate studentship from King’s College London. Additional support was received from the Medical Research Council, UK.

