All published articles of this journal are available on ScienceDirect.

Coexistence of Systemic Sclerosis and Sarcoidosis in a Silica-Exposed Patient: Clinical Insights and Literature Review
Authors Info & Affiliations
Abstract
Background
Systemic sclerosis (SSc) and sarcoidosis are distinct autoimmune diseases with overlapping clinical features, and their coexistence is rare. Both diseases share pathophysiological mechanisms that can be influenced by environmental factors, such as silica exposure, which may contribute to more aggressive forms of systemic sclerosis and increased prevalence of sarcoidosis.
Case Presentation
We, herein, present the case of a 38-year-old Brazilian male exposed to silica and diagnosed with an overlap syndrome of systemic sclerosis and pulmonary sarcoidosis, and treatment with mycophenolate mofetil and prednisone was initiated. Despite initial improvement, disease recurrence occurred during steroid tapering, leading to the addition of rituximab. Over time, the patient showed substantial clinical improvement, with normalization of inflammatory markers and return to normal activities.
Discussion
The association between systemic sclerosis (SSc) and sarcoidosis involves complex immunopathological, environmental, and genetic interactions that trigger similar autoimmune responses. In SSc, silica activates the innate immune system, disrupting immune tolerance and promoting autoantibody production and tissue damage. In sarcoidosis, silica is frequently found in granulomas, suggesting its direct role in the disease's pathogenesis.
Conclusion
This case has highlighted the complexity of diagnosing and managing overlap syndromes, particularly when environmental exposures are involved, and emphasized the importance of a comprehensive diagnostic approach to guide treatment decisions.
1. INTRODUCTION
Systemic sclerosis (SSc) is a rare autoimmune disease characterized by fibrosis and vasculopathy of the skin and internal organs [1, 2]. Sarcoidosis, on the other hand, is an inflammatory disease characterized by the presence of granulomas at various sites, with a wide range of clinical phenotypes [3]. Silica exposure is a common environmental factor that can influence silicosis, sarcoidosis, and systemic sclerosis, with epidemiological studies indicating a higher risk of systemic autoimmune diseases among individuals in high-risk occupations, such as construction work, mining, and masonry, thereby highlighting the importance of occupational health policies [4].
Silica exposure may be associated with a more aggressive phenotype of systemic sclerosis, including diffuse cutaneous involvement and interstitial lung disease. Additionally, silica exposure has been linked to an increased prevalence of sarcoidosis [5-7]. Although distinct, systemic sclerosis (SSc) and sarcoidosis share several pathophysiological characteristics, with multisystem disorders marked by immunological dysregulation and inflammation, which can lead to overlapping clinical manifestations [8]. This paper has presented an association that is rarely described in the literature, i.e., systemic sclerosis and sarcoidosis in a patient exposed to silica.
2. CLINICAL CASE PRESENTATION
A 38-year-old Brazilian male patient sought rheumatological care in the United States (US), reporting significant weight loss of 13 kg over four months, episodes of fever up to 38.3°C, and arthralgia in the hands and ankles. He worked in the construction sector in the US, specifically in the manufacturing and installation of interlocking pavers, having frequent exposure to silica dust for years. He also reported moderate exertional dyspnea associated with a dry cough. He had previously been diagnosed with bacterial pneumonia and treated at another medical facility, but showed no clinical improvement. The patient denied a family history of rheumatological diseases, unprotected sexual activity, recent vaccination, contact with sick individuals, smoking, alcohol consumption, illicit drug use, or exposure to particulate matter, diesel exhaust, or heavy metals. On physical examination, the patient weighed 67.5 kg, having a blood pressure of 112/80 mmHg, a heart rate of 103 bpm, and a temperature of 37.8°C. Pulse oximetry was not measurable. Rheumatological evaluation revealed skin thickening over the metacarpophalangeal joints, forearms, thoracic region, and quadriceps (Rodnan score: 21), active Raynaud’s phenomenon in hands, ulcerations and stellate scars on the distal phalanges, and nasal thinning and telangiectasias on the palms and chest. Joint examination demonstrated synovitis in the wrists and ankles, with pain and localized warmth. Abdominal examination showed a soft and non-tender abdomen without visceromegaly, and no palpable lymphadenopathy. Lung auscultation revealed crackles at the lung bases, and oxygen saturation could not be measured reliably due to active Raynaud’s phenomenon, which impaired peripheral pulse oximetry readings.
Given the clinical manifestations suggestive of systemic sclerosis, a comprehensive diagnostic workup was initiated, including the exclusion of potential infectious causes, to facilitate early treatment initiation. The main diagnostic test results are summarized in Table 1.
| Hemoglobin: 11.3 g/dL | ESR: 80mm/h |
| Leukocytes: 11760/mm3 | CRP: 93.9 mg/L |
| Lymphocytes: 1176/mm3 | ANA: 1:640, homogeneous nuclear pattern |
| Creatinine: 0.7 mg/dL | Anti-Ro: Positive |
| Urea: 20 mg/dL | Anti-La: Not reactive (NR) |
| AST: 64 U/L | Anti-Sm: Not reactive (NR) |
| ALT: 65 U/L | Anti-RNP: Not reactive (NR) |
| CPK: 1.504 U/L | C3: 167 mg/dL |
| C4: 48 mg/dL |
Laboratory tests showed a positive anti-SCL70 antibody, while anti-RNA polymerase III and anticentromere antibodies were negative. Serologies for hepatitis B and C, HIV, syphilis, and IGRA were negative. The periungual capillaroscopy showed an SD (scleroderma pattern) (Fig. 1).
Although there were no parenchymal changes on chest computed tomography (CT), there was mediastinal lymphadenopathy and multiple pulmonary nodules, some calcified, predominantly in the upper lobes (Fig. 2). Spirometry revealed a moderate restrictive ventilatory disorder, with forced vital capacity (FVC) of 2.93 L (63%), forced expiratory volume in 1 second (FEV1) of 2.35 L (62%), and the ratio of FEV1 to FVC (FEV1/FVC) of 81 (98%), with no response to a bronchodilator. The assessment of DLCO (diffusing capacity of the lung for carbon monoxide) was not performed. The transthoracic echocardiogram showed atrial and ventricular chambers with normal volumes and thicknesses, normally functioning valves, and a pulmonary artery systolic pressure of 27 mmHg. Signs of erosive esophagitis and mild antral gastritis were also found on the endoscopic examination.
Clinical, laboratory, and imaging findings were consistent with a diagnosis of diffuse cutaneous systemic sclerosis, which scored 17 points in the ACR/EULAR 2013 classification criteria.

Periungual capillaroscopy showing intense capillary dilation and devascularization, compatible with the scleroderma pattern (SD).
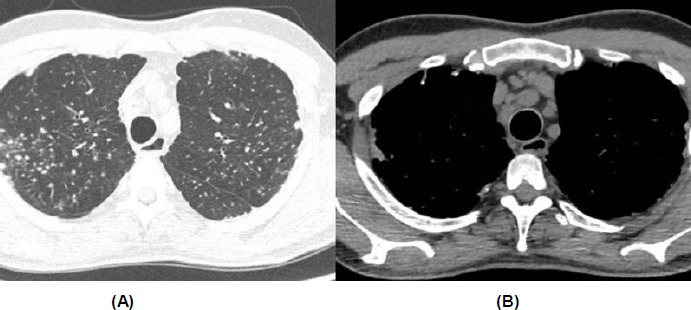
Chest computed tomography. (A) Presence of small, non-specific pulmonary nodules. (B) Increased number of lymph nodes and mediastinal lymphadenopathy.
However, the tomography did not show typical parenchymal changes associated with interstitial lung diseases related to systemic sclerosis, and it revealed diffuse mediastinal lymphadenopathy. As a result, a pulmonology evaluation was requested, especially considering the patient's significant occupational exposure to silica, which could be an important differential diagnosis. At this moment, the main hypotheses raised for the case were sarcoidosis, silicosis, or miliary tuberculosis. Given the clinical picture, a bronchoscopy with transbronchial biopsy was recommended.
The histopathological examination revealed non-necrotizing granulomatous inflammation with areas of hyalinization, findings consistent with sarcoidosis. Stains for bacilli and fungi, including Kinyoun, GMS, and PAS, were negative, excluding the possibility of tuberculosis or any other infectious process (Fig. 3). Polarized light microscopy identified a small quantity of material consistent with silica, insufficient to corroborate the diagnosis of silicosis associated with systemic sclerosis.
After the biopsy result, the diagnosis of overlap of diffuse cutaneous systemic sclerosis without definitive evidence of pulmonary involvement, associated with pulmonary sarcoidosis, was established in a patient with a history of silica exposure. At this time, mycophenolate mofetil 1g/day was initiated. After the diagnosis and the start of treatment, the patient had to temporarily return to Brazil, seeking rheumatology care again. After reviewing the case, the decision was made to increase the mycophenolate dose to the full dose of 3g/day, and due to the intense symptoms more related to sarcoidosis, it was decided to start prednisone at 20mg/day, under surveillance due to the risk of scleroderma renal crisis.
After three months, there was a significant improvement in synovitis, myositis, inflammation, and respiratory symptoms. However, during gradual steroid dose reduction, disease recurrence occurred, evidenced by wrist edema and elevated inflammatory markers (ESR 59 mm/h and CRP 43.1 mg/dL). Due to significant cutaneous involvement and recurrence of joint symptoms associated with the tapering of corticosteroids, the decision was made to add an infusion of 2g of rituximab. One month after the infusion, with no side effects, the patient was able to return to the United States.
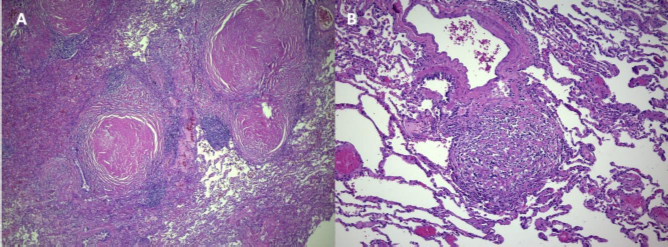
(A) Lymphangitic distribution nodules with extensive hyalinization surrounded by lymphoid aggregates, HE; 4X. (B) Hyalinized nodule with fibroblasts and histiocytes, HE; 20X.
Three months after the first infusion, the patient showed favorable progress, allowing complete discontinuation of corticosteroids. A year and a half after starting treatment, his Rodnan skin score decreased from 21 to 6 points, with recovery of usual body weight and substantial improvement of lymphadenopathy on imaging. Inflammatory markers also improved, with ESR decreasing to 38 mm/h and CRP to 5.9 mg/dL, ultimately allowing the patient to resume normal daily activities without symptoms.
3. METHODOLOGY AND SEARCH STRATEGY
This study involved a descriptive design, combining a clinical case report with a case-based literature review. A qualitative methodology was employed, given the nature of the research question and data sources. The study population consisted of published clinical cases of patients with overlapping systemic sclerosis and sarcoidosis, retrieved from the literature. No experimental or interventional procedures were applied. Therefore, the study was non-experimental, non-observational, and categorized as descriptive with qualitative analysis. The literature search was conducted using the PubMed® and Embase® databases. The following keywords were employed: “systemic sclerosis” and “sarcoidosis”. Only articles published in the English language and providing full access to individual clinical case data were considered. Titles and abstracts were screened for eligibility, and full texts of potentially relevant articles were reviewed. Studies were included if they described one or more cases of patients with both diagnoses, regardless of gender, age, or geographic origin. The variables extracted from each report included the following: patient demographics, age at disease onset, serological markers (autoantibodies), biopsy site for sarcoidosis confirmation, and therapeutic interventions. Data were organized in a comparative table, and key findings were summarized narratively. No statistical analysis was conducted due to the qualitative nature of the study and the small number of available reports.
This methodology allowed for the identification of clinical patterns and knowledge gaps related to this rare overlap syndrome, helping in contextualizing the presented case. A total of 26 studies were selected and are represented in Table 2. The flowchart is presented in (Fig. 4). The ICF (informed consent form) was collected from the patient, and the analysis was conducted following the Helsinki criteria. The English language of the article was improved using ChatGPT.
4. DISCUSSION
The association between systemic sclerosis (SSc) and sarcoidosis is rare, with few cases described in the literature. According to Himmel et al., as of 2020, only around 30 cases had been reported. This coexistence appears to be more frequent in patients with systemic sclerosis than in the general population, although no significant impact on mortality has been observed [8]. The present case has highlighted the diagnostic and therapeutic complexities associated with this combination of diseases, requiring individualized assessment of each manifestation to determine the best treatment at the time.
Continuous exposure to silica, as experienced by the patient, is an environmental factor associated with both diseases. The association between systemic sclerosis and silicosis is also referred to as Erasmus syndrome [34]. In a Brazilian observational study, most patients were male smokers with diffuse cutaneous sclerosis, along with Raynaud's phenomenon, esophageal involvement, and interstitial lung disease, similar to our case [35].
The association between systemic sclerosis and sarcoidosis involves complex immunopathological, environmental, and genetic interactions that can trigger similar autoimmune responses [36]. In SSc, this agent activates the innate immune system, breaking immune tolerance and promoting the production of autoantibodies and tissue damage [37]. Furthermore, it contributes to skin fibrosis, possibly through the HDAC4/Smad2/3 pathway, which regulates fibroblast proliferation and activation [38]. In sarcoidosis, silica has also been frequently found in sarcoid granulomas, suggesting a direct role in the disease's pathogenesis [39].
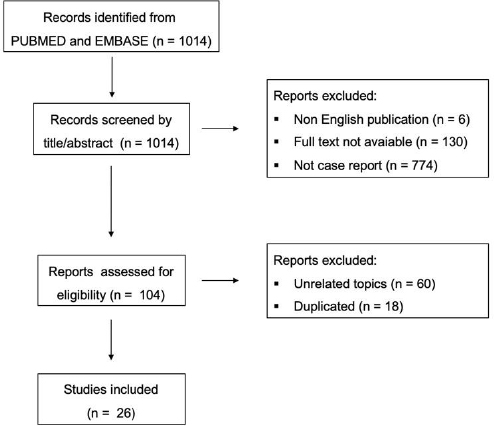
Flowchart for literature search strategy.
| First Author and Year/Refs. | Number of Patients | Gender | Age at Disease Onset | Detected Autoantibodies | Biopsy Site for Sarcoidosis | Initial Treatment |
|---|---|---|---|---|---|---|
| Gogulska et al., 2024 [9] | 3 | 1 male 2 female |
63 76; 69 |
NA | NA | Corticosteroids, methotrexate. and cyclosporine; corticosteroids and methotrexate; corticosteroids, methotrexate, and mycophenolate mofetil |
| Lemay et al., 2023 [10] | 1 | Female | 72 | Antinuclear antibody at 1:2560 (centromere pattern) and 1:160 (speckled), anti-SSA | Lung | Methotrexate 15 mg S/C once per week, prednisone 40 mg/day |
| Reguart Oto et al., 2020 [11] | 1 | Male | 39 | Antinuclear antibody 1:640, anti-Scl-70 | Muscle | NA |
| Nakamura et al., 2023 [12] | 1 | Female | 59 | Antinuclear antibody 1:320 (homogeneous), 1:40 (speckled) | NA | NA |
| Ko et al, 2020 [2] | 1 | Female | 59 | NA | NA | Prednisolone 5mg/day, bucillamine, abatacept |
| Carmi et al., 2018 [13] | 1 | Female | 29 | Antinuclear antibody at 1:160 (speckled), anti-Scl-70 | Lymph node | Corticosteroids |
| Bernardo et al., 2018 [14] | 1 | Female | 62 | Autoantibody screening was negative | NA | Conservative treatment |
| Yu et al., 2017 [15] | 1 | Male | 52 | Antinuclear antibody 1:640 (centromere pattern), 1:160 (speckled), rheumatoid factor, anti-SSA, anti-SSB, and anti-centromere | Lung | Mycophenolate mofetil 500 mg twice daily |
| Yamasue et al., 2017 [16] | 1 | Female | 64 | Antinuclear antibody 1:640, anti-RNA polymerase III | Lung | Prednisolone 25 mg/day and intravenous cyclophosphamide 500 mg |
| Koguchi et al, 2016 [17] | 1 | Female | 75 | Antinuclear antibody 1:1280, anti-centromere | Lung | Conservative and symptomatic treatment |
| Kato et al., 2015 [18] | 1 | Female | 74 | Antinuclear antibody 1:1280, anti-centromere | Skin | NA |
| Senda et al., 2014 [19] | 1 | Female | 59 | Antinuclear antibody 1:1280, anti-centromere | NA | Conservative treatment |
| Ren et al., 2013 [20] | 1 | Female | 54 | NA | Skin | Prednisone 30mg/day, hydroxychloroquine 200mg, and thalidomide 50mg |
| Kobak et al, 2013 [21] | 1 | Female | 52 | Antinuclear antibody 1:320 (nucleolar and homogeneous) | Skin | Corticosteroids, hydroxychloroquine 200 mg/day, azathioprine 150 mg/day |
| Mahajan et al., 2012 [22] | 1 | Female | 41 | Antinuclear antibody 1:40 (speckled), anti-centromere | Skin | Hydroxychloroquine 400 mg/day, prednisolone 40 mg/day |
| Ogane et al., 2012 [23] | 1 | Female | 65 | Antinuclear antibodies 1:160 (homogeneous), anti-Scl-70 | Skin, kidney | Prednisolone 40 mg/day |
| Yamamoto et al., 2012 [24] | 1 | Female | 71 | Antinuclear antibody 1:2560 (speckled), anti-DNA, anti-centromere | Skin | NA |
| Suga et al., 2011 [25] | 1 | Female | 59 | Antinuclear antibody 1:320 (speckled), anti-U1 RNP | Muscle | Prednisone 30 mg/day |
| Sakamoto et al., 2010 [26] | 1 | Female | 73 | Antinuclear antibody 1:640, anticentromere | Skin | NA |
| da Costa et al., 2009 [27] | 1 | Male | 45 | Antinuclear antibody 1:640 (antinucleolar), anti-Scl 70 | Lung | Prednisone 30mg/day |
| Lis-Swiety et al., 2006 [28] | 1 | Female | 17 | Antinuclear antibodies, anti-Ku, anti-smooth muscle antibodies, rheumatoid factor, anticardiolipin | Lung | Prednisolone 40 mg/day |
| Tillon et al., 2006 [29] | 1 | Female | 61 | Antinuclear antibody 1:1200, anti-centromere | Skin | Conservative and symptomatic treatment |
| Takahashy et al., 1997 [30] | 1 | Female | 46 | Antinuclear antibody (homogeneous and speckled), anti-SS-A | NA | Corticosteroids 0.5 mg/kg/day |
| Hosoya et al., 1995 [31] | 1 | Female | 56 | Antinuclear antibody 1:40 (speckled), rheumatoid factor, anti-SSA, anti-SSB | Lymph node | Prednisolone 60mg |
| Maekawa et al. [32], | 1 | Female | 50 | Antinuclear antibody 1:160, anti-centromere | NA | Prednisolone 30mg |
| Groen et al., 1993 [33] | 1 | Male | 44 | Antinuclear antibody 1:40, anti-scl-70 | Lung, muscle, lymph node | Prednisolone 40mg |
The underlying pathophysiological mechanisms of overlap syndromes, especially in systemic sclerosis and sarcoidosis, remain poorly understood. Recent genomic studies have demonstrated that distinct autoimmune diseases often share common susceptibility loci. A large-scale cross-disease genetic analysis involving more than 129,000 individuals showed approximately 40% of genetic associations across autoimmune conditions to be driven by shared alleles, supporting the existence of common pathogenic predispositions between clinically distinct diseases, such as SSc and sarcoidosis [40]. In the context of systemic sclerosis, several genes have been associated with disease susceptibility, including STAT4, IRF5, TNFAIP3, FCGR/FCRL, and PLD4, many of which are implicated in other autoimmune disorders, such as systemic lupus erythematosus and rheumatoid arthritis. The STAT4 intronic polymorphism, in particular, has been strongly associated with hyperresponsive immune phenotypes and sustained inflammatory activation [41].
These findings suggest that certain genetic profiles may confer increased risk for multiple or overlapping autoimmune syndromes. Twin studies have further supported this hypothesis, showing a high concordance for autoantibody profiles (e.g., ANA and anti-Scl-70) among monozygotic twins, but relatively low concordance for the clinical manifestation of systemic sclerosis itself. This implies that while genetic factors influence the immunologic signature, environmental exposures are likely crucial for triggering clinical disease expression [42]. Sarcoidosis also exhibits familial aggregation and heritability, although its associated genetic loci remain less clearly defined [3].
In the absence of other identifiable triggers, the documented occupational exposure to silica may represent a relevant environmental factor, potentially contributing to the development of this rare overlap, although causality cannot be established. Although the amount of birefringent particles identified under polarized light microscopy was small and insufficient for a histopathologic diagnosis of silicosis, their presence reinforced the hypothesis of a gene-environment interaction as a possible driver of the overlap syndrome observed in the patient in the presented case [41].
Moreover, increasing evidence suggests that exposure to inorganic particles, such as silica, may induce epigenetic modifications, including changes in DNA methylation and microRNA expression. These epigenetic alterations can affect fibrotic and immune-regulatory pathways, such as those involving collagen genes, the TGF-β signaling axis, and WNT pathways, thereby amplifying autoimmunity and fibrosis in genetically predisposed individuals [43]. Such mechanisms may help explain the emergence of complex and overlapping autoimmune phenotypes, as illustrated by this case [44].
Despite meeting SSc criteria and having high-resolution CT, typically sensitive and specific for interstitial lung disease (ILD), the patient’s atypical imaging and differential diagnoses of silicosis and tuberculosis justified histopathological confirmation, as recommended by the European Respiratory Society (ERS) [45]. With the results showing no signs of silicosis and a negative tuberculosis culture, the presence of non-necrotizing granulomas, coupled with systemic prodromal symptoms, like fever, weight loss, and arthritis, led to a concurrent diagnosis of sarcoidosis.
In a case series, six patients presented with concomitant scleroderma and sarcoidosis. Symptoms, such as fever, weight loss, and respiratory issues, were common. Radiographs showed intrathoracic lymphadenopathy and/or interstitial lung disease, and lung or lymph node biopsies revealed non-caseating granulomas. Treatment with corticosteroids improved symptoms in these patients [46]. Although corticosteroids are not recommended due to the risk of scleroderma renal crisis, they remain the first-line treatment for inflammation control in sarcoidosis [3]. Therefore, in our case, corticosteroids were used as the initial therapy due to significantly elevated inflammatory markers, with careful monitoring for complications.
Among the 33 cases reviewed in Table 2, there was observed a clear predominance of female patients (26 out of 33), with a mean age at disease onset of approximately 56.6 years. Antinuclear antibody (ANA) positivity was a nearly universal finding, with frequent identification of anti-centromere, anti-Scl-70, and anti-SSA/SSB autoantibodies. Sarcoidosis was most commonly confirmed via biopsy of the skin (10 cases), lung (7 cases), lymph nodes (3 cases), or muscle (3 cases). Corticosteroids were the mainstay of initial treatment in most cases, either as monotherapy or in combination with immunosuppressants. Only a minority of patients were managed conservatively.
Our case shared several clinical, immunological, and therapeutic similarities with those reported in the literature. The patient had high-titer ANA (1:640) and tested positive for anti-Scl-70 antibodies, features commonly seen in other reports [11, 13, 23, 27]. Systemic inflammatory symptoms, such as fever, weight loss, arthritis, and elevated inflammatory markers, were also prominent in our case, aligning with typical overlap presentations. The use of corticosteroids combined with mycophenolate mofetil in the initial management was consistent with many prior cases. Additionally, the case presented by Reguart Oto et al. [11] was noteworthy for its inclusion of a male patient with anti-Scl-70 positivity and concomitant exposure to silica, making it the only other report that explicitly documented an environmental trigger of relevance. A review of the literature showed most patients with concomitant scleroderma and sarcoidosis positive for anti-Scl-70 to be men, while those positive for anticentromere antibodies to be mostly women [19]. A similar result was observed in our case, as the patient was male and anti-Scl-70 positive, suggesting a possible relationship between autoantibody profiles and sex with respect to the coexistence of scleroderma and sarcoidosis.
However, our case also presented notable distinctions. The patient was a 38-year-old male, significantly younger than the average reported age, and exhibited diffuse cutaneous systemic sclerosis, a phenotype less frequently documented. Although environmental exposure to silica has not been mentioned in most reviewed cases, the presented patient’s occupational history and the presence of birefringent particles under polarized light microscopy supported silica as a plausible trigger, despite the amount being insufficient for a formal diagnosis of silicosis. Therapeutically, the addition of rituximab due to disease recurrence during corticosteroid tapering represented a deviation from the standard approach, as rituximab is not commonly used. Given the limited evidence for rituximab in sarcoidosis, restricted to case reports and small series, its combination with mycophenolate mofetil has been guided by its established efficacy in systemic sclerosis, where both agents benefit skin and lung involvement [47-49]. Mycophenolate has also been reported to serve as a steroid-sparing agent in sarcoidosis, with retrospective studies supporting its role in chronic pulmonary and neurologic forms of the disease [50-52]. These clinical and therapeutic aspects have highlighted the uniqueness and complexity of the present case within the broader spectrum of systemic sclerosis-sarcoidosis overlap.
CONCLUSION
The coexistence of systemic sclerosis and sarcoidosis is a rare phenomenon, but it has significant clinical implications, especially when associated with environmental silica exposure. This case has illustrated the diagnostic and therapeutic challenges inherent in recognizing overlap syndromes. Early identification of the overlap between these two diseases is crucial, as they share similar immunopathological mechanisms but require distinct therapeutic strategies to control their systemic manifestations. This case has emphasized the need for continuous monitoring and careful assessment of environmental exposures when considering diagnosis and treatment choices. Further studies are warranted to better understand the pathogenesis and interplay between these diseases, particularly the role of gene-environment interactions in autoimmune overlap syndromes, and define the optimal use of tailored immunosuppressive therapies based on dominant clinical features and disease progression.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: G.P.C., G.C.P., V.F.A: Study conception and design; N.P.M., F.F.M.G., R.M.P., D.M.S., P.C.S: Data collection. All authors have reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| ALT | = Alanine aminotransferase |
| ANA | = Antinuclear antibody |
| AST | = Aspartate aminotransferase |
| CK | = Creatine phosphokinase |
| CRP | = C-reactive protein |
| CT | = Computed tomography |
| DLCO | = Diffusing capacity of the lung for carbon monoxide |
| ESR | = Erythrocyte sedimentation rate |
| ERS | = European Respiratory Society |
| FEV1 | = Forced expiratory volume in 1 second |
| FEV1/FVC | = FEV1 to forced vital capacity ratio |
| FVC | = Forced vital capacity |
| GMS | = Gomori methenamine silver stain |
| HE | = Hematoxylin and eosin |
| HIV | = Human immunodeficiency virus |
| IGRA | = Interferon-gamma release assay |
| ICF | = Informed consent form |
| PAS | = Periodic acid–Schiff stain |
| SD | = Scleroderma pattern |
| SSc | = Systemic sclerosis |
| US | = United States |
| Ur | = Urea |
ETHICAL STATEMENT
This study signed the approved informed consent form from the ethics committee of the Hospital de Clínicas da Universidade Federal do Paraná (CHC-UFPR), Brazil, under protocol number CAAE 39133220700000096.
CONSENT FOR PUBLICATION
Informed consent was obtained from the patient for the publication of this case report.
AVAILABILITY OF DATA AND MATERIALS
The data used in this study will be available from the corresponding author [G.C.P] upon reasonable request.
ACKNOWLEDGEMENTS
Declared none.

