All published articles of this journal are available on ScienceDirect.

First-in-human Study to Evaluate a Single Injection of KiOmedine®CM-Chitosan for Treating Symptomatic Knee Osteoarthritis
Authors Info & Affiliations
Abstract
Background:
Single-injection viscosupplementation is currently performed with cross-linked hyaluronan (e.g., Durolane®) for treating symptomatic knee osteoarthritis.
Objective:
This first-in-human study evaluated the safety and performance of single-injection treatment with non-crosslinked KiOmedine®CM-Chitosan.
Methods:
Patients with painful knee osteoarthritis were randomly assigned to the KiOmedine®CM-Chitosan (n=63) or Durolane® (n=32) group. Patients were blinded to treatment and followed up for 26 weeks. Durolane® was used as scientific control to ensure the validity of the study and reliability of results. No direct comparison was performed between the two groups. The primary objective was defined as an intra-group effect size of 0.8 at 13 weeks post-injection compared to baseline on WOMAC-A (pain). Secondary outcomes included self-reported knee stiffness and knee function, responder rate, quality-of-life questionnaires, and safety.
Results:
The primary objective for both the KiOmedine®CM-Chitosan and the Durolane® groups was met: mean pain reduction of 62.5% (effect size 2.08) for the KiOmedine®CM-Chitosan group and 62.4% (effect size 2.28) for the Durolane® group. Secondary performance outcomes showed all clinically relevant treatment effects over 26 weeks for both groups (p<0.05). Treatment-related adverse events were more often reported in the KiOmedine®CM-Chitosan than Durolane® group and were limited to local reactions. No serious treatment-related adverse events were reported.
Conclusion:
A single intra-articular injection of non-crosslinked KiOmedine®CM-Chitosan is safe and effective for treating symptomatic knee osteoarthritis with a high responder rate. Pain reduction is maintained for 6 months with a high responder rate.
The clinical trial registration number: NCT03679208.
1. INTRODUCTION
Osteoarthritis is the most common chronic musculoskeletal condition [1] and is characterized by joint pain and dysfunction due to progressive subchondral bone damage, articular cartilage loss, inflammation/synovitis and osteophyte formation [2]. Advancing age is the main risk factor for developing osteoarthritis, along with risk factors such as obesity, genetic predisposition, low bone density, trauma, and gender [1]. Knee osteoarthritis is present in approximately 30% of individuals over the age of 65 years and is a major cause of disability in the elderly [1].
Synovial fluid acts as a joint lubricant during shear stress and a shock absorber during compressive stress, and its viscosity is primarily determined by endogenous hyaluronan [3]. Knee osteoarthritis reduces the concentration and molecular weight of endogenous hyaluronan, resulting in the reduction of viscoelastic properties of the synovial fluid. Consequently, the knee joint is less protected from shear and compressive stress, and pro-inflammatory pathways become activated [4-6]. The therapeutic approach for treating knee osteoarthritis consists of drugs with pharmacological actions, such as pain-relief drugs, and other non-pharmacological treatments, such as viscosupplementation. Viscosupplementation is the intra-articular injection of a viscous/elastic solution or gel to counteract the loss of lubrication of synovial fluid due to osteoarthritis [7].
Single-injection treatments have now become the standard as opposed to multi-injection methods. Available CE-marked single-injection viscosupplementation devices are all composed of chemically cross-linked hyaluronan [8]. New viscosupplementation devices based on new molecules are now becoming available on the market. One of them is the new fluid biomaterial implant: KiOmedine®CM-Chitosan,. It is composed of 2.0% non-crosslinked chitosan derivative of non-animal origin, obtained by proprietary chemistry after extraction from the edible white mushroom, Agaricus bisporus. It is a single-injection treatment. A 3-mL volume of KiOmedine®CM-Chitosan is suitable for intra-articular injection into the knee in order to provide synovial joint lubrication [9]. Compared to the cross-linked hyaluronan, KiOmedine®CM-Chitosan has a higher lubrication capacity and a higher ability to fight against oxidative stress [10].
The main objective of this first-in-human study is to evaluate the clinical efficacy and safety of a single intra-articular injection of 3-mL KiOmedine®CM-Chitosan implant for the treatment of symptomatic knee osteoarthritis. This is not a non-inferiority study, and the viscosupplement Durolane® was selected only as a scientific control group rather than comparing its safety and efficacy to KiOmedine®CM-Chitosan. Similar to KiOmedine®CM-Chitosan, it requires a single-injection regimen and a 3-mL dose.
2. MATERIALS AND METHODS
2.1. Materials
KiOmedine®CM-chitosan was obtained by controlled derivatization following the method previously described [11, 12]. A minimum of 100 g of chitosan, extracted from Agaricus bisporus, was dispersed in a reaction medium composed of isopropanol and sodium hydroxide. Monochloracetic acid was added to the chitosan suspension. The obtained carboxymethyl chitosan was subsequently reacted with acetic anhydride, and extensively purified by precipitation in ethanol. The precipitate was dried out to remove excess solvent and residual water, to yield the polymer. KiOmedine®CM-chitosan was formulated at 1.8-2.2% (w:w) in phosphate-buffered water for injection at pH 7.2±0.2 supplemented with 3.5% sorbitol (w:w). The biomaterial was filled in a glass syringe and steam sterilized. All of the raw materials and excipients conformed to European Pharmacopoeia standards for injectable use.
2.2. Study Design
This study was a first-in-human prospective, multicentre cohort with a 26-week follow-up period and was initiated in 2018. The study was single-blinded, meaning that the patient was blinded for the type of injected product. The study monitor (Factory CRO at Bilthoven, The Netherlands) ensured that this clinical investigation was conducted, recorded, and reported in accordance with good clinical practices and data protection regulations, specifically, the Clinical Investigation Plan; standard operating procedures and ISO 14155:2011; clinical investigation of medical devices for human subjects - Good clinical practice, and the requirements of the Declaration of Helsinki; or with applicable country-specific regulatory requirements - whichever afforded greater protection to the subject. Favourable opinion/approval was obtained from the National Competent Authority and Central Ethics Committees in the targeted countries. No patient was included before signing the consent form. Results are presented in line with the guideline from the International Committee of Medical Journal Editors (ICMJE) [13].
2.3. Participants, Eligibility Criteria, and Settings
The eligibility criteria of study participants were defined in accordance with the current medical practice in viscosupplementation and were consistent with the population of patients who were enrolled in clinical trials in viscosupplementation. Patients were informed of the study design by the investigator, as per good clinical practice and ISO14155, before signing the consent form. Male and female patients were recruited who were suffering from symptomatic pain in the treatment knee for at least 26 weeks, and either were not responding or responding poorly to simple oral analgesics (non-opioid analgesics and non-steroidal anti-inflammatory drugs). Patients aged 40–85 years had a body mass index (BMI) ≤ 35 kg/m2. In addition, patients had to be diagnosed with knee osteoarthritis according to the clinical and radiological criteria of the American College of Rheumatology [14] with a Kellgren & Lawrence grade II to III [15]; patients needed to have a pain score of 7-17 at baseline, when assessed with the 5-graded Likert WOMAC A after a mandatory 48-hour washout period of medication and minimal contralateral knee pain (i.e., WOMAC A ≤ 6).
Exclusion criteria were Radiological Kellgren & Lawrence grade 0, I or IV; exclusively patellofemoral osteoarthritis; chondromatosis or villonodular synovitis of the knee; clinically-apparent knee effusion, inflammation, or flare-up of the knee or abnormal synovial fluid macroscopy or significant volume upon arthrocentesis on the day of injection; history of injury or trauma in the treatment knee; significant clinically-assessed or radiographic varus or valgus deformation; inflammatory disease or pathologies interfering with the evaluation of osteoarthritis pain including homolateral coxarthrosis; hyaluronan injection, arthroscopy, or surgery in the treatment knee in the 26 weeks prior to injection; oral corticotherapy ≥5 mg/day, injection of corticosteroids or platelet-rich plasma or cell-based therapy in the 13 weeks prior to injection; change in the dosage regimen of symptomatic slow-acting drugs or in physiotherapy in the 13 weeks prior to injection; Anticipated need for any surgery or any forbidden OA treatments; history of recurrent infection or synovial infection; hypersensitivity or allergy to the product components of KIO014; history of symptomatic hip osteoarthritis; history of autoimmune disease; any severe, ongoing, and uncontrolled disease or any investigator-assessed clinically significant condition that may represent a substantial risk to the patient or impact study assessments; alcohol addiction; sever alteration of mobility; high risk of haemorrhage; participation in a therapeutic clinical trial in the last 13 weeks before injection; pregnancy or breastfeeding; being under guardianship or judicial protection.
2.4. Interventions
This study was a first-in-human trial to investigate the safety and efficacy of the treatment with KiOmedine®CM-Chitosan (KiOmed Pharma) in patients with symptomatic knee osteoarthritis. Durolane® (Bioventus) was used as a control to support the reliability and scientific validity of the clinical evaluation procedure for the efficacy and safety of KiOmedine®CM-Chitosan. Both Durolane® and KiOmedine®CM-Chitosan can be used with a single-injection of the 3-mL dose and can be injected with the same type of syringe.
The assessment schedule consisted of a screening visit, a baseline visit for injecting KiOmedine®CM-Chitosan or Durolane®, and four follow-up visits at 2, 6, 13, and 26 weeks post-injection. For each visit, patients were asked not to take any pain medication in the last 48 hours before the visit (i.e., 48-hour washout period). The unauthorized use of rescue medication and/or non-respect for the 48-hour washout were handled as protocol deviations in the analysis populations.
All injections and assessments were performed by qualified physicians with experience in intra-articular injections and clinical research in the relevant medical field. Accurate needle placement into the synovial joint was confirmed by slight aspiration of synovial fluid at the time of the intra-articular injection. Before intra-articular injection, the target knee was carefully examined. and synovial fluid was aspirated as needed.
2.5. Outcomes (Primary and Secondary)
The primary efficacy objective was to demonstrate that, compared to baseline, the intra-group effect size on pain is ≥ 0.8 at 13 weeks after a single injection of KiOmedine®CM-Chitosan. An effect size of 0.8 on pain at 13 weeks post-injection is considered clinically large per Cohen’s threshold, is consistent with a clinically relevant effect on pain reported for viscosupplementation with hyaluronan [16], and is superior to the consensus intra-articular placebo effect of 0.6 [17]. In addition, the effect size is a common way of measuring the magnitude of the treatment effect in clinical studies on knee pain in patients with osteoarthritis [16, 18]. Secondary study outcomes for the different time points (baseline and 2-, 6-, 13-, and 26-weeks post-injection) included self-reported pain, self-reported functional health and well-being, responder rate, patients’ satisfaction, physician’s satisfaction from a medical and usability point-of-view, and adverse events.
Clinical assessments included a Self-administered WOMAC questionnaire for pain, stiffness and function [19-21]; a Self-administered questionnaire for pain and global assessment (scale of 0 to 10) [22]; Osteoarthritis Research Society International (OARSI) Standing Committee for Clinical Trials Response Criteria Initiative and the Outcome Measures in Rheumatology (OMERACT)” responder criteria for osteoarthritis clinical trials [22] to evaluate response to treatment; Self-administered SF-12 health survey of functional health and well-being (scale of 0 to 100) [23]; and five-point Likert scales for the usability of the investigational device. Consistent with the primary efficacy endpoint used in clinical studies reported for Durolane® [24, 25], the responder rate was defined as a reduction in the WOMAC pain score of at least 40% with an absolute improvement of at least five points compared with baseline for the treatment knee at the different visits [23]. Pre-injection pain (baseline WOMAC score) forms the basis for showing efficacy as intended, meaning that the patient is serving as his or her own control. For OMERACT-OARSI responder criteria, an improvement in pain or in function ≥ 50% and an absolute change ≥ 20 were considered as a positive response to treatment.
Of all adverse events, the investigator recorded the incidence, severity (i.e., mild, moderate, severe), seriousness, and the relationship to the study treatment or device. Safety of the investigational medical device was evaluated at the different time points by recording the incidence, severity, and causal relationship of (serious) adverse events, (serious) adverse device effects, unanticipated serious adverse device effects and device deficiencies. Local effects such as joint pain (arthralgia), joint effusion, joint swelling, joint warmth, injection site pain, and joint stiffness are anticipated with viscosupplementation in the treatment knee. The severity and occurrence of each local effect were assessed using a four-point numerical rating scale (none, mild, moderate, serious).
2.6. Sample Size Calculation
2.6.1. Sample Size
In this randomized controlled cohort, 60 patients were planned to receive a single intra-articular injection of KiOmedine®CM-Chitosan, while the other 30 patients were planned to receive a single intra-articular injection of the control Durolane®. In the KiOmedine®CM-Chitosan group, the sample size of 60 patients was determined to ensure an accurate assessment of the single injection efficacy on pain, considering a consensus intra-group (before/after injection) effect size of intra-articular placebo of 0.6 at 13 weeks post-injection [17]. The sample size assumed a dropout rate of 10%. With a minimum of 54 completed patients, the study could show 99% power to demonstrate any statistically significant reduction in baseline pain with a significance level of 5% for any effect size ≥ 0.6. For the Durolane® group, a sample size of 31 patients was deemed sufficient to demonstrate any significant reduction in pre-injection baseline pain with appropriate power (99%) and alpha (5%) if the intra-group effect size of the treatment is at least 0.8 at 13 weeks post-injection. This effect size is consistent with clinical trials and meta-analyses on single-injection viscosupplementation, including on Durolane® [24-27].
For safety evaluations, the study was designed to have 90% power to detect at least one adverse event of interest or more if its true rate of occurrence is approximately 3%. This is consistent with the evaluation of treatment-related adverse event incidence of local effects reported from the single-injection hyaluronan benchmarks [24, 25, 28, 29].
2.7. Interim Analyses and Stopping Guidelines
Interim analysis was done after 30 patients who received a single injection of KiOmedine®CM-Chitosan completed Visit 4 (3 months post-injection).
Trial was designed to be terminated by the Sponsor for any of the following reasons: protocol deviations, device deficiency or malfunction, safety concern, production limitation, administrative decisions. Recommendation for termination or modification of the trial for excessive adverse events was at the discretion of the Data Monitoring Committee.
2.8. Randomization (Random Number Generation, Allocation Concealment, Implementation)
Randomization in 2:1 allocation to the KiOmedine®CM-Chitosan and Durolane® groups, respectively, was achieved via a computer-generated program, which also stratified qualified subjects by study sites. Sealed envelopes with sequential numbers, containing randomization codes were provided to the study sites and were used in sequential order. Patients were not informed of their randomization assignment until the end of the study.
2.9. Blinding
Each patient was blinded for the type of treatment at the injection visit (single blinding). Both Durolane® and KiOmedine®CM-Chitosan can be injected with the same type of syringe; This allows blind delivery of the product to the patient. KiOmedine®CM-Chitosan and Durolane® were supplied in a sterile and ready-to-use 3-mL syringe in identical packages labelled with a unique identification code. Breaking the single blinding only occurred in the case of major device deficiency or safety concerns (repeated SADEs) upon Sponsor’s decision, and/or in any circumstance that was deemed necessary for the safeguard of those patients.
2.10. Statistical Analyses
Durolane® was used as a control treatment to validate the clinical evaluation methods used in this trial. The outcomes of this control group were not intended for comparing the two treatments. The primary analysis population for efficacy and safety was the ‘Safety Population’, which was constituted of all patients who received any randomized study treatment with at least one follow-up visit. Missing values were not replaced nor extrapolated. All statistical analyses were performed using IBM SPSS Statistics (Version 21.0).
Descriptive statistical analyses were performed using mean, median, standard deviation (SD), minimum and maximum values for continuous variables, and using sample size (n), absolute (n) and relative incidence (%) for discrete variables. Inferential statistical analyses were performed with a significance level of 5% and 90% power or using non-parametric rank tests. Where appropriate, Bonferroni’s correction for multiplicity was applied to ensure the trial was conclusive for several efficacy parameters and to provide a stringent control of Type I errors (i.e., minimizing the chance of a false positive discovery).
For the primary efficacy endpoint, any change in pre-injection baseline pain at 13 weeks was evaluated using paired Student’s t-test. The intra-group effect size was calculated as the difference between mean WOMAC A (pain) at baseline and mean WOMAC A at 13 weeks divided by the pooled standard deviation from the two data sets, assuming a normal distribution of data. The over-time modification of the efficacy endpoints was assessed versus baseline by analyses of variance for repeated measures, followed by appropriate post-hoc tests, for continuous variables with a normal distribution. For discrete variables and for continuous variables for which the assumption of normality was not fulfilled, Friedman’s tests were performed, followed when significant by 2 by 2 Wilcoxon’s tests.
For safety analysis, all recorded adverse events were coded using version 21.0 of the MedDRA and were summarized by presenting the number and percentage of patients showing any sign or symptom. Post-injection adverse events were analysed in three categories based on the causality assessment: treatment-emergent adverse events irrespective of their causality with the procedure and the device; treatment-related adverse events with a possible, probable, or causal relationship with the procedure and/or the device; and adverse device effects with a possible, probable, or causal relationship with the device only. Safety analysis was subject to the independent Data Monitoring Committee’s (DMC) review scheduled per protocol. DMC is composed of one pharmacologist/ biostatistician, one orthopaedist and one general physician.
3. RESULTS
3.1. Demographics and Baseline Characteristics
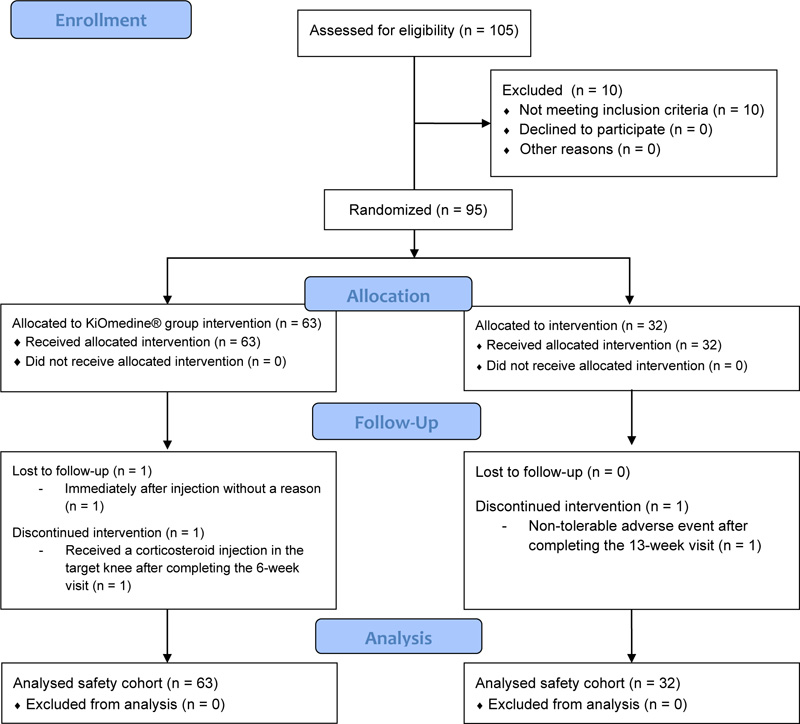
Ten (10) patients were screen failures (i.e., patient who signed the informed consent but did not meet the eligibility criteria at either the screening or injection visit). A total of 95 patients were enrolled, from 11 March 2019 to 03 June 2019, and randomly assigned to the study treatments, of which 63 patients were injected once with KiOmedine®CM-Chitosan and 32 patients were injected once with Durolane® (Fig. 1). The randomization ratio between KiOmedine®CM-Chitosan and Durolane® was 1.97 instead of 2.00. In the KiOmedine®CM-Chitosan group, 23.8% (n=15) of patients were recruited in four centers in the Netherlands and 76.2% (n=48) in three centers in Hungary. In the Durolane® group, 25% (n=8) of patients were recruited in the Netherlands and 75% (n=24) in Hungary. There were no protocol deviations, thus all randomized patients were included in the safety cohort for analysis. Results from the per-protocol cohort, constituting of all patients who adhered to the protocol and attended all visits up to 13 weeks post-injection, are provided in supplemental materials. The per-protocol cohort includes 51 patients in the KiOmedine®CM-Chitosan group and 28 patients in the Durolane® group.
There were no significant differences in demographics and baseline characteristics between the KiOmedine®CM-Chitosan and Durolane® group, except that more males were injected in the Durolane® group (Fisher’s exact test, p = 0.040;) (Table 1). This difference was not considered clinically significant for the purpose of this study.
| Characteristics |
KiOmedine®CM-Chitosan (n = 63) |
Durolane® (n = 32) |
|---|---|---|
| Age in years, mean (SD) | 61.9 (8.3) | 59.8 (8.1) |
| Gender | ||
| Female, n (%) | 47 (74.6) | 17 (53.1)† |
| Male, n (%) | 16 (25.4) | 15 (46.9)† |
| Race, n (%) | ||
| White/Caucasians | 63 (100) | 32 (100) |
| BMI in kg/m2, mean (SD) | 28.9 (3.2) | 29.1 (3.3) |
| Disease duration in years, mean (SD) | 4.8 (6.0) | 5.1 (5.7) |
| Kellgren-Lawrence, n (%) | ||
| Grade II | 39 (61.9) | 21 (65.6) |
| Grade III | 24 (38.1) | 11 (34.4) |
| Other osteoarthritis sites, n (%)# | 38 (60.3) | 19 (59.4) |
| Contralateral knee osteoarthritis | 32 (50.8) | 16 (50.0) |
| Of which symptomatic knee osteoarthritis$ | 4 (6.2) | 2 (6.3) |
| Hip osteoarthritis | 3 (4.8) | 1 (3.1) |
| Synovial fluid aspirated prior to injection, n (%) | 39 (61.9) | 20 (62.5) |
| Injection position, n (%) | ||
| Anterolateral | 24 (38.1) | 12 (37.5) |
| Lateral mid patellar | 17 (27.0) | 8 (25.0) |
| Lateral suprapatellar | 22 (34.9) | 12 (37.5) |
| WOMAC Total [0-96], mean (SD) | 51.4 (15.5) | 51.7 (14.4) |
| WOMAC A Pain [0-20], mean (SD) | 11.2 (2.5) | 11.7 (2.4) |
| 48-hour washout, n (%) | 62 (98.4) | 32 (100) |
The intra-articular position of the injection was anterolateral, lateral mid patellar, or lateral suprapatellar. Of note, the skin at the injection site was considered in good condition in all patients, except for one patient with poor condition of the local skin. However, the injection site was acceptable for intra-articular injection.
3.2. Primary Efficacy Outcome
In the KiOmedine®CM-Chitosan group, WOMAC A (pain) score at 13 weeks post-injection decreased by 62.5% from baseline (11.2 ±2.5 at baseline vs. 4.2 ±4.0 at 13 weeks post-injection; paired Student’s t test, p < 0.001). The effect size equalled 2.08. In the Durolane® group, there was a 62.4% reduction in WOMAC A (pain) score at 13 weeks post-injection compared to baseline (11.7 ±2.4 at baseline vs. 4.4 ±3.8 at 13 weeks post-injection; paired Student’s t test, p < 0.001). The effects size equalled 2.28.
3.3. Safety Outcomes
The overall incidence of adverse events was comparable between KiOmedine®CM-Chitosan (n = 27, 42.9%) and Durolane® (n = 13, 40.6%) (Table 2). There was one serious adverse event (1.6%). This SAE consisted of biliary colic, which was not related to the device and was resolved with hospitalization.
In the KiOmedine®CM-Chitosan group, 23 patients (36.5%) experienced at least one treatment-emergent adverse event occurring in the treatment knee, 19 patients (30.2%) experienced a treatment-related adverse event, and nine patients (14.3%) experienced adverse device effect. Their severity was considered mild to moderate in most cases. Meanwhile, one adverse device effect was considered severe in one patient. No treatment-related adverse event or adverse device effect led to study withdrawal. The most commonly reported treatment-related adverse event was arthralgia (25.4%) in the treatment knee. Other treatment-related adverse events were joint effusion, joint swelling, or synovitis. A concomitant medication was prescribed in 85.7% of cases, and a concurrent procedure consisting of synovial fluid aspiration was performed in two cases. The most relevant adverse events occurred within one day after injection (mean: 0.2 day; median: 0 day) and it was resolved within 2-3 weeks (mean: 16.5 days; median: 3 days). No long-term onset adverse events were recorded.
| Incidence, n (%) | KiOmedine®CM-Chitosan (n=63) | Durolane®(n=32) |
|---|---|---|
| Patients with at least one AE | 27 (42.9) | 13 (40.6) |
| Patients with at least one TEAE | 23 (36.5) | 7 (21.9) |
| Patients with at least one TRAE*$ | 19 (30.2) | 4 (12.5) |
| Arthralgia | 16 (25.4) | 3 (9.4) |
| Joint effusion | 4 (6.3) | 0 (0.0) |
| Joint swelling | 4 (6.3) | 0 (0.0) |
| Synovitis | 3 (4.8) | 1 (3.1) |
| Discharge | 1 (1.6) | 0 (0.0) |
| Musculoskeletal stiffness | 0 (0.0) | 1 (3.1) |
| Patients with at least one ADE* | 9 (14.3) | 1 (3.1) |
3.4. Secondary Efficacy Outcome
3.4.1. Self-administered WOMAC Questionnaire for Pain, Stiffness, and Function
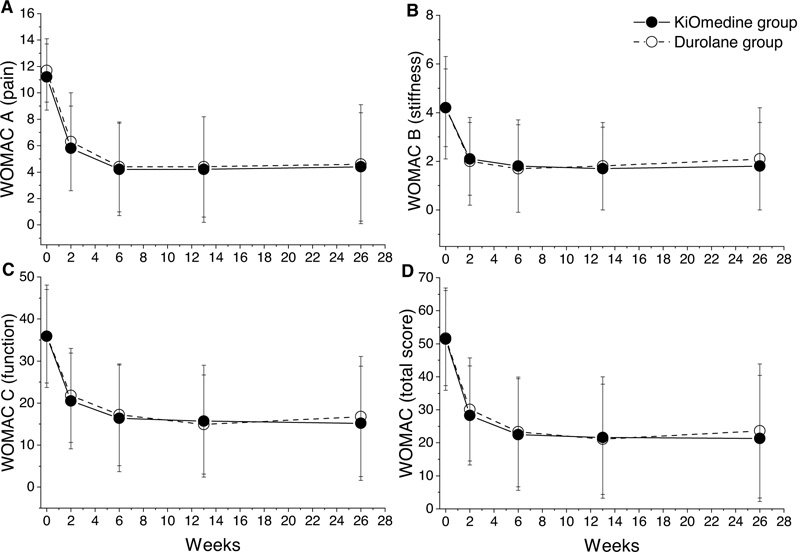
The repeated measures analysis of variance for WOMAC pain (Fig. 2A), WOMAC stiffness (Fig. 2B), and WOMAC function (Fig. 2C) revealed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (p < 0.001 for all comparisons) (Table 3). Post-hoc Bonferroni’s tests revealed a statistically significant reduction in WOMAC pain and WOMAC function scores between baseline and all further time points (all p < 0.001) and between week 2 and all further time points (all p < 0.005). Moreover, the WOMAC stiffness score significantly decreased from baseline to all further time points (Post-hoc Bonferroni’s tests, all p < 0.001).
| Variables | Randomization | Day 0 | Week 2 | Week 6 | Week 13 | Week 26 |
|---|---|---|---|---|---|---|
| Sample size, n | KiOmedine® CM-Chitosan | 63 | 62 | 62 | 60 | 61 |
| - | Durolane ® | 32 | 32 | 32 | 32 | 31 |
| WOMAC A (pain) | KiOmedine® CM-Chitosan | 11.2 (2.5) | 5.8 (3.2)† | 4.2 (3.5)† | 4.2 (4.0)† | 4.4 (4.1)† |
| - | Durolane® | 11.7 (2.4) | 6.3 (3.7)† | 4.4 (3.4)† | 4.4 (3.8)† | 4.6 (4.5)† |
| WOMAC B (stiffness) | KiOmedine® CM-Chitosan | 4.2 (1.6) | 2.1 (1.5)† | 1.8 (1.9)† | 1.7 (1.7)† | 1.8 (1.8)† |
| - | Durolane® | 4.2 (2.1) | 2.0 (1.8)† | 1.7 (1.8)† | 1.8 (1.8)† | 2.1 (2.1)† |
| WOMAC C (function) | KiOmedine® CM-Chitosan | 35.9 (12.2) | 20.5 (11.4)† | 16.4 (12.7)† | 15.7 (13.3)† | 15.2 (13.6)† |
| - | Durolane® | 35.9 (11.1) | 21.8 (11.2)† | 17.2 (12.1)† | 14.9 (11.8)† | 16.8 (14.3)† |
| WOMAC (total) | KiOmedine® CM-Chitosan | 51.4 (15.5) | 28.3 (15.0)† | 22.5 (16.9)† | 21.6 (18.4)† | 21.3 (19.1)† |
| - | Durolane® | 51.7 (14.4) | 30.1 (15.6)† | 23.3 (16.6)† | 21.1 (16.7)† | 23.6 (20.3)† |
| Pain at rest | KiOmedine® CM-Chitosan | 6.3 (1.8)* | 3.1 (2.1)† | 2.0 (2.3)† | 1.8 (2.2)† | 2.0 (2.3)† |
| - | Durolane® | 6.2 (1.5) | 3.2 (2.1)† | 2.0 (2.0)† | 1.9 (2.2)† | 1.8 (2.3)† |
| Patients’ global assessment | KiOmedine® CM-Chitosan | 6.8 (1.4)* | 3.4 (2.1)† | 2.7 (2.4)† | 2.3 (2.4)† | 2.4 (2.3)† |
| - | Durolane® | 6.7 (1.3) | 3.8 (1.8)† | 2.5 (2.0)† | 2.6 (2.2)† | 2.2 (2.6)† |
| OMERACT-OARSI responders | KiOmedine® CM-Chitosan | N/A | N/A | N/A | 64.5% | 69.4% |
| - | Durolane® | N/A | N/A | N/A | 75.0% | 65.6% |
| WOMAC A (pain) responders | KiOmedine® CM-Chitosan | N/A | 59.7% | 77.4% | 76.7% | 75.4% |
| - | Durolane® | N/A | 56.3% | 84.4% | 84.4% | 71.1% |
| SF-12 physical component | KiOmedine® CM-Chitosan | 33.0 (6.8) | 41.7 (7.4)† | 44.0 (8.2)† | 45.0 (9.3)† | 44.9 (8.6)† |
| - | Durolane® | 33.9 (5.2) | 40.0 (6.3)† | 43.4 (7.3)† | 45.2 (9.1)† | 44.9 (10.2)† |
| SF-12 mental component | KiOmedine® CM-Chitosan | 47.8 (10.4) | 50.7 (8.8)† | 54.9 (8.9)† | 55.1 (7.8)† | 53.4 (8.0)† |
| - | Durolane® | 48.9 (7.7) | 52.7 (8.3)† | 55.5 (7.2)† | 52.6 (10.8)† | 53.7 (8.9)† |
The repeated measures analysis of variance for WOMAC total revealed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p < 0.001) Table 3 and Fig. 2D). Post-hoc Bonferroni’s tests revealed a statistically significant reduction in WOMAC total score between baseline and all further time points (all p < 0.001) and between week 2 and all further time points (all p < 0.005). Additionally, at month 3 and at month 6, there were no statistically significant differences between KiOmedine® CM-chitosan and Durolane® for all WOMAC scores.
3.4.2. Self-administered Questionnaire for Pain and Global Assessment
The repeated measures analysis of variance for both pain and the global assessment score showed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p < 0.001) (Table 3). Post-hoc Bonferroni’s tests revealed a statistically significant reduction in both pain and the global assessment score between baseline and all further time points (all p < 0.001) and between week 2 and all further time points (all p < 0.005).
3.4.3. Responders to Treatment
3.4.3.1. OMERACT-OARSI Responders
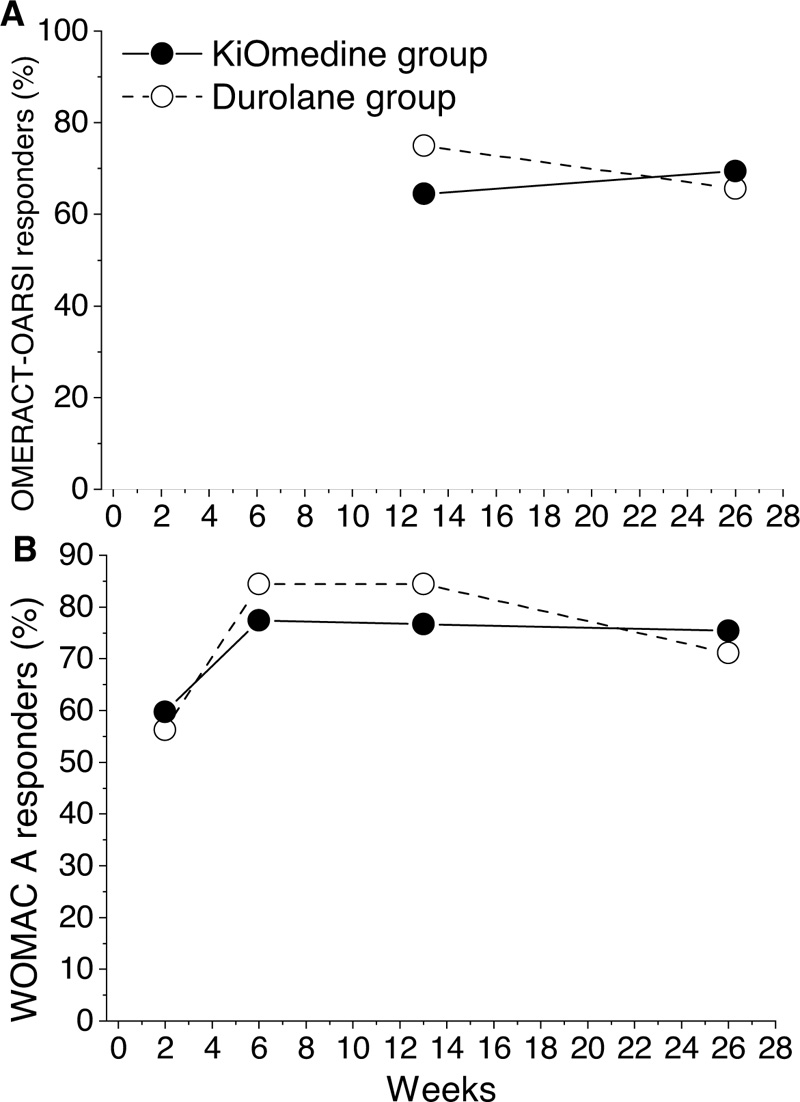
The number and percentage of patients that responded to the treatment have been determined at 13 and 26 weeks post-injection in line with the OMERACT-OARSI [30]. For the KiOmedine®CM-Chitosan group, the percentage of responders was 64.5% (n = 40) at 13 weeks post-injection and 69.4% (n = 43) at 26 weeks post-injection. For the Durolane® group, the percentage of responders was 75.0% (n = 24) at 13 weeks post-injection and 65.6% (n = 21) at 26 weeks post-injection (Table 3 and Fig. 3A). Additionally, at month 3 and at month 6, there was no statistically significant differences between KiOmedine®CM-chitosan and Durolane®.
3.4.3.2. Responders in Terms of WOMAC A (Pain) Change
For the KiOmedine®CM-Chitosan group, the percentage of responders was 76.7% (n = 46) at 13 weeks post-injection, and was maintained at 75.4% (n = 46) at 26 weeks post-injection. For the Durolane® group, the percentage of responders was 84.4% (n = 27) at 13 weeks post-injection, and 71.1% (n = 22) at 26 weeks post-injection (Table 3 and Fig. 3B). Additionally, at month 3 and at month 6, there were no statistically significant differences between KiOmedine®CM-chitosan and Durolane®
3.4.4. Self-administered SF-12 Health Survey of Functional Health and Well-being
The repeated measures analysis of variance for both functional health and well-being showed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p < 0.001) (Table 3). Improvements in functional health between baseline and all further time points were significant (Post-hoc Bonferroni’s tests, p < 0.001 for all comparisons) and between week 2 and all further time points (p < 0.005 for all comparisons). Improvement in well-being between baseline and all further time points (Bonferroni’s tests, p < 0.05 for all comparisons) and between week 2 and week 6 were significant (Bonferroni’s tests, both p < 0.001). Additionally, at month 3 and at month 6, there were no statistically significant differences between KiOmedine®CM-chitosan and Durolane®.
3.4.5. Patient and Physician Satisfaction
3.4.5.1. Patient Satisfaction
The repeated measures analysis of variance for patient satisfaction showed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p = 0.008). Post-hoc Bonferroni’s tests showed a statistically significant increase in patient satisfaction between week 2 and week 13 post-injection and between week 2 and week 26 post-injection (all p ≤ 0.027). Overall, most patients in both groups reported to be satisfied or very satisfied post-injection (82.0% in the KiOmedine®CM-Chitosan group and 74.2% in the Durolane® group at week 26) (Table 4).
| - | KiOmedine®CM-Chitosan | Durolane® | ||
|---|---|---|---|---|
| - | Satisfied or Very Satisfied | Unsatisfied or Very Unsatisfied | Satisfied or Very Satisfied | Unsatisfied or Very Unsatisfied |
| Patient satisfaction | 82.0% (n = 50) | 11.5% (n = 7) | 74.2% (n = 23) | 3.2% (n = 1) |
| Patient’s reported improvement in health | 67.2% (n = 41) | 6.6% (n = 4) | 71.0% (n = 22) | 3.2% (n = 1) |
| Physician satisfaction with knee pain reduction | 82.0% of patients (n = 50) | 6.6% of patients (n = 4) | 74.2% of patients (n = 23) | 9.7% of patients (n = 3) |
| Physician satisfaction with the improvement in knee function | 80.3% of patients (n = 49) | 4.9% of patients (n = 3) | 80.6% of patients (n = 25) | 6.5% of patients (n = 2) |
| Physician satisfaction with the improvement in patient’s health | 73.8% of patients (n = 45) | 4.9% of patients (n = 3) | 74.2% of patients (n = 23) | 6.5% of patients (n = 2) |
| Ease of Injection | Very Easy to Inject | Slight Pressure | Moderate Pressure | Strong Pressure with Light Pain | Impossible to Inject |
|---|---|---|---|---|---|
| KiOmedine® CM-Chitosan | 20 (31.7%) | 42 (66.7%) | 1 (1.6%) | 0 (0.0%) | 0 (0.0%) |
| Durolane® | 0 (0.0%) | 9 (28.1%) | 14 (43.8%) | 9 (28.1%) | 0 (0.0%) |
| Usability assessment: | Very good | Good | Neutral | Poor | Very poor |
| KiOmedine®CM-Chitosan | 34 (54.0%) | 17 (27.0%) | 12 (19.0%) | 0 (0.0%) | 0 (0.0%) |
| Durolane® | 6 (18.8%) | 16 (50.0%) | 8 (25.0%) | 2 (6.3%)* | 0 (0.0%) |
The repeated measures analysis of variance for patient’s health revealed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p < 0.001). Post-hoc Bonferroni’s tests showed a statistically significant improvement in patient’s health between week 2 and all further time points (p < 0.005 for all comparisons). Overall, most patients in both groups reported that their health was improved or very much improved post-injection (67.2% in the KiOmedine® CM-Chitosan group and 71.0% in the Durolane® group at week 26) (Table 4). Additionally, at month 3 and at month 6, there were no statistically significant differences between KiOmedine®CM-chitosan and Durolane®.
3.4.5.2. Physician Satisfaction
The repeated measures analysis of variance for the clinical evaluation of knee pain showed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p = 0.024). The clinical evaluation of knee pain showed a significant improvement from week 2 to week 13 post-injection (Post-hoc Bonferroni’s tests, both p = 0.047).
The repeated measures analysis of variance for the clinical evaluation of knee function showed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p = 0.009). The clinical evaluation of knee function showed a significant improvement from week 2 to week 6 post-injection (Post-hoc Bonferroni’s tests; both p = 0.010).
The repeated measures analysis of variance for the clinical evaluation of patient’s health showed a statistically significant effect of time for the KiOmedine®CM-Chitosan and Durolane® groups (both p < 0.001). Patient’s health significantly improved from week 2 to week 6 and from week 2 to week 13 post-injection (Post-hoc Bonferroni’s test, both p < 0.001 for all comparisons). At month 3 and at month 6, there was no statistically significant differences between KiOmedine® CM-chitosan and Durolane®.
Overall, the physicians were satisfied to very satisfied with the improvement in health, knee function, and reduction in knee pain for patients of both treatment methods (Table 4).
3.4.6. Ease of Injection and Usability Assessment
In all patients except one, physicians found KiOmedine®CM-Chitosan very easy to inject or needed only a slight pressure (Table 5). None of the physicians found Durolane® very easy to inject, and for the majority of Durolane® injections, a moderate pressure (43.8%) or strong pressure with light pain (28.1%) was required (Table 5). Furthermore, the overall usability assessment of the device was considered good to very good. No device deficiency was reported for KiOmedine®CM-Chitosan, and two device deficiencies were reported for Durolane® (Table 5). The two device deficiencies for Durolane® were reported to correspond to suboptimal intra-articular volume delivery in relation to high injection force. The comparison was statistically significant for both the ease of injection and the usability assessment. The comfort and usability were better with KiOmedine®CM-chitosan compared to Durolane®.
4. DISCUSSION
This was a prospective cohort study to evaluate the safety and efficacy of a single injection of a non-crosslinked bioimplant, KiOmedine®CM-Chitosan, for treating symptomatic knee osteoarthritis. Our results demonstrated that a single intra-articular injection of KiOmedine®CM-Chitosan obtains a statistically significant and clinically relevant reduction of knee pain. At 13 weeks post-injection, the mean reduction in knee pain was 62.5% compared to baseline; the intra-group effect size in pain reduction was 2.08, larger than the effect size threshold of 0.8. Sustained reduction from baseline pain was maintained over the 26 weeks study period. At 26 weeks post-injection, the reduction of WOMAC pain was 61% from baseline; the treatment responder rate was between 69.4% and 75.4%, depending on the criteria used to define responders. This relative change in baseline pain is considered a substantial clinically important difference (>50%) according to the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) working group [31-33]. This treatment also provided significant improvements in WOMAC function score and the physical and mental health components of the SF-12 in the KiOmedine®CM-Chitosan group for up to 26 weeks. The improvement of >10 points on the patient-related SF-12 physical component largely exceeded the minimal clinically important difference reported for pain relief (4.5 points) and function (4.8 points) [34]. Overall, >80% of patients were satisfied with the treatment at 13 weeks and at 26 weeks. The Durolane® group was included as a scientific control group for regulatory purposes only. The clinical efficacy of Durolane® in our study was considered to be consistent with that reported in clinical studies [24, 25, 35-37].
Although the proportion of patients who experienced at least one adverse event was similar for both treatments, the proportion of patients with at least one treatment-emergent adverse event or treatment-related adverse event was lower for the Durolane® than KiOmedine®CM-Chitosan group (Table 5). Moreover, joint effusion, joint swelling, and discharge were not reported for the Durolane® group. But, in general, KiOmedine®CM-Chitosan was well tolerated in the target population. There was no serious adverse event or patient withdrawal related to the study treatment. Treatment-related adverse events and adverse device effects were local reactions in the treatment knee. The most relevant adverse events were reported as arthralgia, joint effusion, or joint swelling within one-day post-injection. Most reactions were either mild to moderate in intensity and self-limited. Three moderate synovitis reactions were reported that were characterized by painful effusion. All reactions responded well to rest and simple oral analgesics, and they did not adversely affect the clinical efficacy of the treatment. Two of them required a synovial fluid aspiration to remove the volume of fluid built up in excess. Of note, one case of synovitis that required synovial fluid aspiration was also recorded in one patient who was injected with Durolane®. Macroscopically, the aspirated fluid was neither purulent nor bloody, nor did it present signs of significant inflammation. Following aspiration, the patients rapidly improved. This reaction was neither assimilated to a pseudoseptic or anaphylactic reaction nor joint infection. No long-term onset of adverse events was reported at the end of the study. These treatment-related adverse events seem to be induced by a physiological macrophage reaction that is needed to degrade the CM-chitosan implant [9]. This reaction could be exacerbated in pre-reactivated synovial membrane (even in low-grade inflammation osteoarthritis patients) but responded well to conservative concomitant pain medication and had no impact on the clinical benefit.
By comparison with single-injection hyaluronan viscosupplementation products, transient local reactions reported with KiOmedine®CM-Chitosan are common post-injection side effects of viscosupplementation in the target population. For example, the overall incidence of treatment-related adverse events was 35.8% in clinical investigations with Synvisc-One® [26] and 49.0% in clinical investigations with Gel-One® [38], mostly consisting of arthralgia, joint effusion, and joint swelling. In clinical studies with Durolane® [24, 25, 28, 35], the overall incidence of treatment-related adverse events ranged from 12.7% to 18.4%, with arthralgia being the most prominent adverse event. The Durolane® group in the present study demonstrated a similar overall incidence rate of treatment-related adverse events (12.5%), of which the majority was attributed to arthralgia. The KiOmedine®CM-Chitosan group showed a higher incidence rate of treatment-related adverse events (30.2%) than Durolane®, but this incidence rate was still lower compared to other well-known viscosupplementation products such as Synvisc-One® [26] and Gel-One® [38].
This study had some limitations. First, the study objectives and sample size were not powered for non-inferiority against the control Durolane®, which limited any statistical comparison between the treatments used in the randomized controlled cohort. For this reason, the statistical analysis indicated in this manuscript between the two treatments is only indicative. Durolane® was included with the aim of supporting the reliability and scientific validity of the study, particularly as it relates to subjective assessment of pain in an indication affected by numerous confounding factors such as disease progression or placebo effect. Second, only single blinding of patients to the treatment was used. However, single blinding will likely not impact the conclusions of the study because the assessment of clinical efficacy was self-reported by the patient and not the physician.
CONCLUSION
In conclusion, the present study showed that a single intra-articular injection of 3-mL KiOmedine®CM-Chitosan in the treatment of symptomatic knee osteoarthritis resulted in statistically significant and clinically relevant improvements in pain and patient-related function over 26 weeks with a high response rate. Similar performance results were obtained for the Durolane® group. Even if the frequency of the adverse event observed in the KiOmedine® CM-chitosan group is higher than Durolane® group, the treatment is considered safe, compared to other viscosupplementation products on the market. The overall safety profile of KiOmedine®CM-chitosan was found to be acceptable based on an independent DMC’s safety review. Future clinical studies will confirm the long-term (beyond 6 months – study on going) [39] of safety and performance of KiOmedine®CM-chitosan and will be designed to directly compare the efficacy of KiOmedine®CM-Chitosan and cross-linked viscosupplementation products or a placebo in non-inferiority trials. Further studies will also investigate the long-term efficacy following a single injection with KiOmedine®CM-Chitosan in advanced osteoarthritis [40] (study on-going) [41], where the unmet medical need is higher because many of these patients do not respond well to cross-linked hyaluronan-based viscosupplementation.
LIST OF ABBREVIATIONS
| IV | = Intravenous |
| DMC | = Data Monitoring Committee |
| SD | = Standard Deviation |
AUTHORS' CONTRIBUTIONS
Conceptualization, P.D.; methodology, P.D., J.B. and Avania; validation, P.D. and M.C.; formal analysis, J.B. (independent statistician); investigation, P.J.E., G.S. and D.H.; resources, P.D and M.C.; data curation, Avania; writing-original draft preparation, Avania; writing-review and editing, MC., L.H.; M.S. visualization, Avania; supervision, P.D.; project administration, P.D.; funding acquisition, N/A All authors have read and agreed to the published version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the Ethics Committee of Slotervaart Hospital and Reade in the Netherlands (protocol code: NL66642.048.18; date of approval: September 05, 2018) and the Scientific and Research Ethics Committee of the Medical Research Council (MRC-SREC) in Hungary (protocol code: OGYÉI/43406/2018; date of approval: September 05, 2018). Due to unforeseeable circumstances at the Medical Center Slotervaart, unrelated to the study, the ethical dossier was transferred on November 06, 2018 to the medical ethical reviewing committee of the Isala Klinieken Zwolle, as to serve as the central ethics committee in the Netherlands for the duration of the study.
HUMAN AND ANIMAL RIGHTS
No animals were used for studies that are the basis of this research. All the humans used were in accordance with the Helsinki Declaration of 1975.
CONSENT FOR PUBLICATION
Every participating patient had provided signed informed consent according to the regulatory and legal requirements.
STANDARDS OF REPORTING
Consort guidelines were followed.
AVAILABILITY OF DATA AND MATERIALS
The data presented in this study are available on request from the corresponding author [M.C].
FUNDING
This research was funded by the Research and Technological Innovation programs of Biowin, grant number 7360-Prouesse and the Walloon Region, grant number 7842-Kiogel and “The APC was funded by Kiomed Pharma”.
CONFLICT OF INTEREST
All the institutions of the approved study group received funding from KiOmed Pharma to support the research reported in this paper. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Dr. Gábor Skaliczki reports no conflicts of interest that could impact on the research. Dr. Daniel Haverkamp reports no conflicts of interest that could impact on the research. Dr. Peter Emans is a paid consultant for KiOmed Pharma. Dr. Jacques Bentin is a paid consultant for KiOmed Pharma. Pierre Douette was a full-time consultant for KiOmed Pharma. Mickaël Chausson is a full-time employee of KiOmed Pharma. Laurence Hermitte was a full-time consultant for KiOmed Pharma.
ACKNOWLEDGEMENTS
The APROOVE study group: Dr. Daniel Haverkamp (national study coordinator), Dr. Boudijn Joling, Dr. Harm van der Vis, Dr. Daniël Hoornenborg, Amsterdam, The Netherlands; Dr. Rutger Zuurmond, Dr. Annemarie Brandsma, Zwolle, The Netherlands Dr. Dirk Das, Dr. Rogier van Drumpt), Geldrop, The Netherlands; Dr. Pieter Emans, Dr. Tim Boymans, Maastricht, The Netherlands ; Dr. Gábor Skaliczki (national study coordinator), Dr. Imre Sallai, Budapest, Hungary ; Dr. György Gruber, Dr. László Tajti, Mako, Hungary; Dr. Endre Lénárt, Dr. Katalin Faragó (in memoriam), Dr. Katalin Töröcsik, Kecskemét, Hungary; Statistics: Jacques Bruwhyler, Linkebeek, Belgium.
While the content of the manuscript was developed by the authors, the actual development of the manuscript was facilitated by Azar Kordbacheh, Tjerk Zult and Federica Colonna from a third-party company called Avania.
SUPPORTIVE/SUPPLEMENTARY MATERIAL (IF ANY):
Efficacy and safety results of Per Protocol (PP) cohort are provided in supplemental materials.

