All published articles of this journal are available on ScienceDirect.

Vitamin D and Anti-Phospholipid Antibody Syndrome: A Comprehensive Review
Authors Info & Affiliations
Abstract
Vitamin D is a steroid hormone that exerts a variety of biological effects that range from the well-known regulation of bone metabolism to the modulation of cellular growth, of apoptosis, and the regulation of both innate and adaptive immunity. Evidence supports a correlation between low vitamin D levels and a high risk to develop chronic inflammatory diseases including autoimmune diseases.
Anti-phospholipid antibody (aPL) syndrome (APS) is an autoimmune chronic condition characterized by recurrent arterial and/or venous thrombosis and/or obstetric complications associated with persistent aPL positivity. Secondary prevention of thrombosis is widely accepted in these patients and relies on life-long anticoagulant drugs. On the contrary, primary prevention in isolated aPL positivity in healthy carriers and treatment of obstetric manifestations in APS are still debated.
Epidemiological data have shown that vitamin D deficiency (serum levels <30 ng/ml) is frequent in APS patients and that it may be associated with an increased risk of thrombosis in these patients. Experimental data show that vitamin D is able to reduce the expression of adhesion molecules, of toll-like receptors and the secretion of proinflammatory chemokines, thus playing a protective role on endothelial activation and the subsequent development of thrombosis in APS.
Although these observations need to be confirmed in prospective studies and randomized clinical trials, it is tempting to speculate that vitamin D supplementation could be very useful for the prevention of clinical manifestations in APS patients, in particular as a primary prevention countermeasure in aPL carriers.
1. INTRODUCTION
Vitamin D is a steroid hormone generated by cholesterol, exerting a wide variety of biological effects that go beyond the well-known role in the regulation of bone metabolism and calcium homeostasis [1, 2]. Vitamin D has been later recognized as responsible for many non-skeletal effects, mainly based on observational studies conducted in large cohorts showing a significant association between low levels of vitamin D (i.e., <20 ng/mL of 25-hydroxy-vitamin D) and the risk of developing cardiovascular (CV), metabolic, neoplastic and autoimmune diseases [3-6].
The biological activity of the active form of vitamin D is triggered by its interaction with the high-affinity Vitamin D Receptor (VDR), which belongs to the superfamily of nuclear hormone receptors [7, 8]. After interaction with its ligand, VDR behaves as a transcription factor binding another receptor, Retinoid X Receptor (RXR), and thus generating a heterodimeric complex that recognizes specific DNA sequences. The VDR is also found in caveoles, invaginations of the plasma membrane with high concentrations of sphingolipids and steroids. Vitamin D binds its specific receptor also at this level, but the transduction pathway of this signal is currently unknown [9]. Activation of VDR induces not only osteometabolic responses, but also a wide variety of biological effects, such as modulation of cellular growth, proliferation, apoptosis, and immune cell activation [10]. Vitamin D regulates both innate and adaptive immunity by means of VDR, which is present in almost all immune cells [2, 11-13]. Furthermore, the presence of VDR allelic polymorphisms has been associated with a major susceptibility to develop an autoimmune disease such as Systemic Lupus Erythematosus (SLE) and Rheumatoid Arthritis (RA) [14, 15].
1.1. Immunomodulatory Effects of Vitamin D
Evidence speaks in favor of a regulatory role of vitamin D both for innate and adaptive immunity [16, 17]. VDR is constitutively expressed in Antigen Presenting Cells (APC), such as macrophages and Dendritic Cells (DC), and is inducible in activated T lymphocytes [11, 18]. Vitamin D promotes a balance of innate and adaptive immunity [19] by regulating molecules related to immune activation such as MHC class II and CD40 among others and the production of Interleukin (IL)-1β, IL-6, IL-12, Tumor Necrosis Factor (TNF)-α, and Macrophage-Colony Stimulating Factor (M-CSF) [20-22]. In addition, vitamin D receptor agonists favor DC proliferation and tolerance [16].
Furthermore, vitamin D can modulate Toll Like Receptor (TLR) response to bacterial infections, with an immunosuppressive effect due to the reduction of the expression of TLR2 and TLR4 on monocytes/macrophages [23-25]. Moreover, vitamin D inhibits lipopolysaccharide (LPS)-induced TNF-α production in a concentration-dependent manner [26-28]. Vitamin D is capable of inhibiting LPS-mediated production of various inflammatory chemokines and cytokines and TLR4 and 5 signaling in myometrial smooth muscle cells; in contrast, IL-10 and TLR10 expression increases in response to treatment with vitamin D, suggesting an inhibitory role in infection-related inflammation [29]. In addition, VDR is expressed also on trophoblast cells, which are influenced by the locally synthesized 1,25(OH)2D, either in an autocrine or paracrine way [30-32]. The local synthesis of active vitamin D seems to play a key role in placental innate immunity regulation [30]. In addition, there is evidence that vitamin D exerts an anti-inflammatory role on trophoblast cells isolated both from normal and preeclampsia subjects [33, 34].
1.2. Anti-Phospholipid Antibody Syndrome (APS)
APS is characterized by arterial or venous thrombosis and/or pregnancy morbidity with persistent positivity of aPL detected by means of three formal diagnostic assays: anti-cardiolipin (aCL), anti-β2glycoprotein I (anti-β2GPI) and Lupus Anticoagulant (LA) [35-37]. The reactivity with the LA assay is mainly mediated by antibodies directed against prothrombin and β2GPI, whereas aCL positivity is mainly caused by β2GPI-dependent aPL. Despite persistent positivity of aPL antibodies, thrombotic events in patients with APS occur occasionally. Based on this clinical observation, a “two hit” hypothesis has been proposed in which the antibody (representing the first hit) induces a thrombophilic state, and the presence of another thrombophilic condition provides the second hit, triggering clotting [36].
Immunoglobulin G (IgG) from APS patients are able to induce endothelial dysfunction, monocyte and platelet activation, as well as overexpression of Tissue Factor (TF), adhesion molecules, and proinflammatory cytokines through activation of TLR4 [38, 39]. There is evidence that aPL may also activate endothelial cells via TLR2- and TLR4-mediated signaling [40, 41]. LPS is the natural ligand of TLR4 and experimental data demonstrate that LPS increases the expression of β2GPI in vascular tissues and triggers aPL-mediated thrombosis [42]. It is well known that the second hit can be provided by several triggers, such as surgery, inflammatory and infectious processes [36, 43]. In particular, the combination of a second hit plus the perturbation of endothelial cells mediated by aPL may overcome the threshold for triggering thrombosis [38, 41].
It is widely recognized that pathogenic mechanisms in obstetric APS are different from those involved in thrombotic manifestations. Intraplacental thrombosis, with subsequent impairment of maternal-fetal blood exchange, was initially suggested as the main pathogenic mechanism of aPL-induced fetal loss and heparin was introduced in the treatment of pregnant APS patients based on this assumption. Indeed, in vitro studies have shown that aPL may induce a procoagulant state at the placental level through several mechanisms [44, 45]. However, it has become clear that thrombotic mechanisms do not fully explain recurrent fetal loss in these patients, and proinflammatory rather than prothrombotic mechanisms have been advocated, such as complement activation, inhibition of trophoblast proliferation, differentiation and migration, defective angiogenesis, embryonal toxicity on preimplantation embryo, decreased production of human Chorionic Gonadotropin (hCG) and human placental lactogen, decreased expression of annexin V as well as acute placental inflammation [36, 46-55]. Furthermore, aPL have been shown to specifically destroy trophoblast, inhibit syncytium formation, halt hCG production, and limit trophoblast invasion [56, 57]. The effect of heparin in the management of patients with obstetric APS seems to be based on mechanisms of action other than anticoagulation [58, 59].
The clinical spectrum of APS actually comprises non-criteria manifestations such as APS nephropathy and Central Nervous System (CNS) symptoms (epilepsy and cognitive abnormalities). Also in this case, clinical manifestations are not fully explained by ischemic mechanisms. A direct effect of aPL on glomerular microcirculation and on neuronal cells has been proposed [60, 61].
1.3. Management of APS and Primary Prevention in aPL Carriers
Due to the risk of relapse after the first episode of thrombosis, the management of patients with APS envisages long-term anticoagulation [62]. Bleeding complications may occur as a result of anticoagulation therapy, thrombocytopenia, use of NSAIDs and congenital or acquired hemorrhagic syndromes such as the rare but possible Lupus anticoagulant-hypoprothrombinemia syndrome [63, 64]. Non-pharmacological management of APS is aimed at managing other CV risk factors such as arterial hypertension, obesity (body mass index >30 kg/m2), diabetes mellitus, smoking, active or treated cancer, use of oral contraceptives, underlying systemic autoimmune diseases and genetic hypercoagulable states [65], or venous prothrombotic factors [66].
Isolated positivity of aPL in healthy carriers is often a conundrum for clinicians. Primary prophylaxis in aPL carriers with Low Dose Aspirine (LDA) remains controversial due to the lack of relevant evidence-based data. In particular, it is unclear whether the benefits of LDA in a low-risk population may outweigh the increased risk of bleeding [67]. Based on aPL specificity and isotype, it is possible to stratify patients, with LA and triple positivity bearing the highest risk for a first thrombotic event [68]. In particular, LA raises the risk of thrombosis by approximately 4-fold [69] and aPL IgG isotype is considered clinically more meaningful compared to IgM isotype [70]. Although hydroxychloroquine has been demonstrated to reduce the risk for venous thromboembolism in SLE [71-73], and to have a CV protective effect [74], evidence is controversial for primary prevention of thrombotic events [75]. Also statins have been advocated as effective drugs for primary and secondary prevention of thrombotic events in APS, not only due to their lipid-lowering effect, but also because of their pleiotropic immunomodulatory, anti-inflammatory, and antithrombotic properties [76]. However, in the absence of sound evidence, statins cannot be recommended in APS patients in the absence of hyperlipidemia [58]. Therefore, primary prevention in aPL carriers is still an unmet need.
1.4. Endothelial Perturbation in Anti-Phospholipid Antibody Syndrome
It is widely accepted that anti-β2GPI antibodies, in the presence of β2GPI, can induce endothelial cell perturbation [77, 78]. In the past, our group has demonstrated that anti-β2GPI is able to induce a proinflammatory phenotype with up-regulation of Endothelial-Leukocyte Adhesion Molecule 1 (ELAM-1) and Intercellular Adhesion Molecule 1 (ICAM-1) [42]. Although the precise mechanism of aPL-induced activation of endothelial cells remains to be determined, TLR4 has been demonstrated to play a central role [36, 79]. More recently, other TLRs have been investigated as potentially involved in APS pathogenesis. TLR3 activation via polyinosinic:polycytidylic acid (Poly I:C) seems to enhance LPS/TLR4-induced production of Interferon (IFN) [80]. TLR9 recognizes viral and bacterial DNA, which contain unmethylated CpG dinucleotides. CpG DNA was found to exert potent proinflammatory actions, such as the expression of adhesion molecules, IL-8, and monocyte chemoattractant protein-1 in Human Endothelial Cells (HUVECs), thus facilitating leukocyte trafficking [81, 82]. Evidence suggests that aPL can also directly activate neutrophils, as a consequence of enhanced granule release, oxidative burst, and increase IL-8 production [83, 84]. After activation, adhesion to endothelial cells and transmigration, neutrophils further amplify the inflammatory process. Furthermore, after stimulation within endosomes TLRs activate transcription factors Interferon Regulatory Factor (IRF)3 or IRF7 and induce type I IFNs [85].
1.5. Vitamin D and the Anti-Phospholipid Antibody Syndrome
The role of vitamin D in immune system regulation could contribute to APS pathogenesis. To date, no data are available regarding VDR polymorphisms and APS. Vitamin D deficiency (<10–20 ng/ml) and insufficiency (<30 ng/mL) are relatively common in autoimmune diseases [86], including APS [87]. Vitamin D deficiency has been reported to be significantly correlated with arterial and venous thrombosis as well as with non-criteria APS manifestations such as neurological and ophthalmic manifestations, pulmonary hypertension, livedo reticularis and skin ulcerations in APS patients [88-90]. Similarly, in a large Swedish study an increase of 50% in the risk of venous and arterial thrombosis was observed during winter compared with other seasons, whereas a significantly lower risk of thrombosis was reported in women who were more sun-exposed [91].
Vitamin D is able to suppress the expression of TLRs such as TLR4, which is responsible for the activation of nuclear factor κB and the signaling cascade that ultimately induces a prothrombotic state in endothelial cells by aPL. Reduced expression of TLRs is accompanied by impaired nuclear factor κB translocation to the nucleus and by reduced TLR-dependent signal transduction [63]. Vitamin D can reduce TF expression induced by proinflammatory stimuli such as TNF-α or LPS on monocytes [92, 93].
Vitamin D deficiency has been linked to a three-fold increase in preeclampsia risk [94, 95]. In a cross-sectional study conducted in women with recurrent pregnancy loss, an association between low vitamin D levels and positivity for aPL was found [96]. In particular, during weeks 24-26 of pregnancy, women with low serum levels of vitamin D have significantly increased risk [97].
Vitamin D can inhibit TLR4 signaling in peripheral blood monocytes of pregnant women at risk for preeclampsia, thereby down-regulating inflammatory pathways and reducing the risk of endothelial cell damage [98]. Notably, Gysler et al. found that vitamin D, either alone or in combination with low molecular weight heparin, is able to attenuate the inflammatory response in trophoblast cells after exposure to a monoclonal murine anti-human β2GPI [99].
1.6. Vitamin D and Endothelial Perturbation in Anti-Phospholipid Syndrome
There is evidence that vitamin D is able to directly modulate endothelial perturbation due to its anti-inflammatory effect. Vitamin D was shown to reduce the expression of adhesion molecules such as ELAM-1 and ICAM-1 on endothelial cells [100]. Equils et al. found that pretreatment of human microvessel endothelial cells with vitamin D inhibited LPS-induced activation of transcription factor NF-κB and secretion of IL-8 [101]. Furthermore, a reduction of IL-8 secretion was shown in coronary artery endothelial cells [102]. Despite the well-known role of endothelial cell perturbation in APS, evidence of a direct role of vitamin D on aPL-mediated endothelial cell perturbation is still scarce. Indeed, Agmon-Levin et al reported that vitamin D is a potent inhibitor of the in vitro expression of TF in endothelial cells stimulated by anti-β2GPI antibodies derived from APS patients [88].
With this as background, we investigated the expression of ELAM-1 and ICAM-1, of TLR3 and 9, the secretion of the proinflammatory chemokine IL-8 and the expression of type I IFNs (IFN-α, IFN-β) in cultures of HUVEC pretreated with vitamin D and incubated with inflammatory stimuli such as LPS, TNF-α or anti-β2GPI IgG isolated by APS subjects. After informed consent, we recruited two subjects with high titer of anti-β2GPI and aCL IgG and IgM and with LA activity fulfilling APS 2006 criteria [35]. Polyclonal IgG from five age- and sex-matched healthy subjects were included as controls (HD). Serum IgG were isolated as as previously described [103].
ELAM-1 is one of the main endothelial activation markers whose expression is up-regulated in response to inflammatory stimuli (e.g. LPS and TNF-α) and to anti-β2GPI antibodies [104]. Overnight pre-treatment with vitamin D significantly reduced ELAM-1 expression induced by both inflammatory stimuli (LPS and TNF-α) and anti-β2GPI IgG (p<0.05) in our in vitro model. We observed no changes in ELAM-1 expression in all other experimental conditions (Fig. 1). ICAM-1 is an adhesion molecule whose expression is both constitutive and up-regulated in response to inflammatory stimuli (e.g. LPS and TNF-α) or antibodies to anti-β2GPI [104]. Overnight pre-treatment with vitamin D significantly reduced ICAM-1 transcription induced by inflammatory stimuli (LPS and TNF-α) and anti-β2GPI polyclonal IgG (p<0.05) (data not shown).
IL-8 is a proinflammatory chemokine whose secretion is up-regulated in response to inflammatory stimuli. It is one of the key regulators of leukocyte trafficking/activation in inflammation and it is also involved in tissue injury, fibrosis and angiogenesis [105]. IL-8 secretion increases in response to LPS and aPL [84]. In agreement with previous studies, we found that overnight pre-treatment with vitamin D resulted in a significant reduction of IL-8 concentration in supernatants from HUVEC cultures stimulated with LPS, Poly I:C and aPL IgG (p<0.05) (Fig. 2).
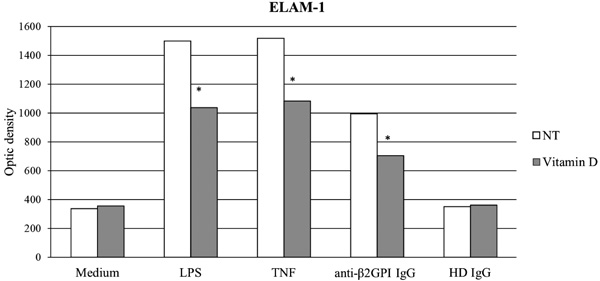
anti-b2GPI: Anti-b2glycoprotein I; HD: Healthy Donor; LPS: lipopolysaccharide; NT: No Treatment; TNF: Tumor Necrosis Factor-alpha. (*p <0.05)
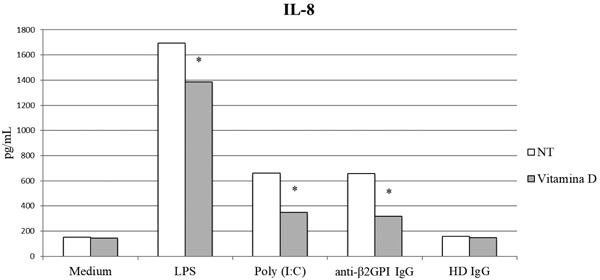
anti-b2GPI: anti-b2glycoprotein I, HD: Healthy Donor, IL-8: Interleukin-8, LPS: Lipopolysaccharide, NT: No Treatment, Poly (I:C): Polyinosinic:polycytidylic acid. (*p <0.05).
Both TLR3 and TLR9 are expressed on the endosomal membranes [85]. Therefore, we studied gene expression of these two receptors. The expression of TLR3 was significantly increased by Poly I:C and LPS, but not by anti-β2GPI, and was reduced by overnight pretreatment with vitamin D (p<0.05). TLR9 was up-regulated by LPS and IgG of patients with APS, but not in the presence of culture medium alone. A slight increase was found in the presence of HD IgG compared to the only culture medium. Overnight pretreatment with vitamin D resulted in increased TLR9 transcription in basal conditions, but significantly reduced receptor expression levels in the presence of LPS, anti-β2GPI IgG (p <0.01) and HD IgG (p <0.05) (data not shown).
Type I IFNs (IFN-α, IFN-β) are important mediators of the immune response that modulate the differentiation and proliferation of immune system cells and contribute to the pathogenesis of several autoimmune diseases [106]. For example, patients with SLE display high and constant levels of IFN-α, responsible for disease activity and organ damage [107, 108]. The analysis of IFN-α and IFN-β mRNA expression showed that pathological Poly I:C, LPS and IgG from APS patients resulted in a significant increase in transcription of both cytokines and a slight increase was also observed in the presence of HD IgG compared to the culture medium alone. The expression of type I IFNs was reduced after overnight pretreatment with vitamin D in all experimental conditions (p<0.01 for LPS and anti-β2GPI IgG; p<0.05 for Poly I:C and HD IgG) (data not shown).
4. DISCUSSION
The therapeutic role of vitamin D supplementation, including the dosage and definition of treatment targets (among which the standardization of the definition of “vitamin D deficiency”) in aPL-positive patients is still to be determined in prospective studies and randomized clinical trials.
Based on the available literature, the administration of vitamin D with a target serum level of vitamin D >30 ng/ml may be beneficial in APS patients. Vitamin D may act as a further countermeasure for secondary prevention in full-blown APS because of its ability to interfere with the prothrombotic mechanisms mediated by aPL. Inflammatory conditions, such as infectious events, may favor the occurrence of the first thrombotic event in asymptomatic aPL carriers [66, 70]. In these patients, vitamin D could provide a potential new pharmacological approach for primary prevention. Finally, its ability to interfere with the mechanisms leading to miscarriages may be useful in preventing clinical manifestations also in obstetric patients. Indeed, vitamin D supplementation should be considered in all APS patients showing vitamin D deficiency and insufficiency [62].
Our experimental data, although preliminary, are in agreement with previous reports. We used an in vitro model of endothelial cell activation with HUVEC stimulated with proinflammatory stimuli - TNF-α and TLR specific agonists such as LPS and Poly I:C - or with anti-β2GPI IgG from APS patients compared with medium culture and HD IgG. We showed that vitamin D can reduce the expression of adhesion molecules such as ELAM-1 and ICAM-1, both expressed as a consequence of an inflammatory stimulus or incubation with aPL IgG [42, 100]. Furthermore, the secretion of IL-8, a proinflammatory chemokine which increases in response to aPL and LPS and is involved in leukocyte recruitment was significantly reduced [84]. TLR9 has been involved in the pathogenesis of APS, whereas TLR3 role has never been investigated in APS pathogenesis [42, 109]. TLR9 was significantly increased both in the presence of inflammatory stimuli and IgG isolated from APS patients and was completely inhibited by pretreatment with vitamin D. Conversely, the expression of TLR3, although reduced by overnight pretreatment with vitamin D, was significantly increased by Poly I:C and LPS, but not by anti-β2GPI. Based on this observation, we assume that TLR9 may be involved in the pathogenesis of APS and could provide a possible target of vitamin D whereas TLR3 may not be directly involved in APS pathogenesis.
TLRs are able to up-regulate expression of type I IFNs; in particular IFN-α transcription is dependent on TLR9 while IFN-β depends on TLR3 and TLR4 activation [110-112]. We observed an increased gene expression of both IFN-α and IFN-β in the presence of both TLR specific agonists and IgG isolated from APS patients and a significant reduction with pretreatment with vitamin D, further confirming its anti-inflammatory effect.
Although our results are preliminary and should be validated with larger experiments and cohort studies, they further support the hypothesis that vitamin D may display a protective effect on endothelial cell perturbation in APS.
CONCLUSION
The anti-inflammatory and antithrombotic role of vitamin D is supported by epidemiological data on vitamin D deficiency in APS subjects, in particular in those with thrombotic manifestations.
It is tempting to speculate that aPL carriers, who do not have an indication for antiplatelet and anticoagulation, may benefit from vitamin D supplementation. Vitamin D may be considered in addition to standard care for obstetric and thrombotic APS patients.
Future mechanistic research should be aimed at investigating the potential effects of vitamin D in APS.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Gruppo LES Italiano provided a grant for RG to run the Lupus Clinic

