All published articles of this journal are available on ScienceDirect.

Carotid Artery Atherosclerosis in Patients with Active Rheumatoid Arthritis: Predictors of Plaque Occurrence and Progression Over 24 Weeks
Abstract
Introduction:
This study evaluated the prevalence and progression of subclinical carotid artery atherosclerosis in active rheumatoid arthritis (RA).
Methods:
Carotid arteries of RA patients were scanned using 3D ultrasound at baseline and 24 weeks for total plaque area, vessel wall volume, and intima-media thickness (IMT), as well as arterial stiffness measured using pulse wave velocity. Variables related to inflammation, lipids and cardiovascular (CV) risk were assessed for associations with plaque progression. Of 195 screened patients, 31 met inclusion criteria (66 Swollen joint count (SJC) plus 68 Tender joint count (TJC)≥8 OR SJC plus TJC≥4 with elevated acute phase reactants) and were enrolled (27 female; mean age 59.3±9.8years). Patients using lipid lowering drugs and uncontrolled comorbidities were excluded.
Results:
Atherosclerotic plaque occurred in 35% and arterial wall hypertrophy (IMT≥0.6mm) in 86% of patients. Most (68%) had an abnormal lipid profile characterized by reduced HDL and/or increased total cholesterol/HDL index, which was adversely affected by disease activity. Stepwise binary logistic regression analysis showed that Framingham risk score (OR=1.155, 95%CI:1.002-1.332, p=0.046) and ESR (OR=1.148, 95%CI:1.015-1.299, p=0.028) predicted plaque burden most strongly. Plaque progression was significantly associated with baseline higher hsCRP, ESR, and heavy smoking, but only hsCRP predicted plaque growth in multivariate regression analysis (p=0.004); and hsCRP was related to higher disease activity (r=0.443, p=0.016), LDL (r=0.544, p=0.007), and smoking (r=0.384, p=0.04).
Conclusion:
RA-related inflammation contributed to augmented CV burden in RA and might mediate its effect on atherosclerosis through hsCRP and modulation of the traditional CV risk factors, such as dyslipidemia.
Keymessages:
- Active rheumatoid arthritis (RA) with long disease duration had a high prevalence of an abnormal lipid bloodprofile, atherosclerotic plaque and carotid artery wall thickness.
- Both traditional CV risk factors and inflammatory disease activity were associated with atherosclerotic burden at baseline and their effects might be interrelated: disease activity had a negative impact on lipid blood profile.
- High-sensitivity CRP serum level was the only significant risk marker for growing atherosclerotic plaque, as measured by 3D ultrasound for total plaque area over 6 months, after adjustment for other covariates.
- The combined effect of traditional cardiovascular risk factors in RA assessed by Framingham risk score failed to predict the high atherosclerotic burden in a quarter of patients; 1.5 multiplication factor recommended for RA patients did not improve risk estimation.
INTRODUCTION
Cardiovascular (CV) death is the leading cause of mortality in patients with RA. It is responsible for approximately half the deaths in RA; CV events occur a decade earlier than in population controls [1], suggesting that, as in diabetes mellitus, RA is a significant independent risk factor for premature ischemic heart disease.
A number of publications have addressed the effects of RA-related risk factors on accelerated atherosclerosis, but most studies were cross-sectional, finding associations, but not necessarily causality [2]. Another limitation of the published observational studies is the choice of carotid intima-media thickness (IMT) as a proxy for cardiovascular diseases. In recent years the role of IMT as a marker of atherosclerosis has been questioned [3, 4]. Thickening of the intima-media layer in common carotid arteries is usually caused by hypertrophy of the smooth muscle cells in the tunica media in response to high shear stress, whereas the atherosclerotic process, particularly in its early phase, is restricted to the intimal layer [5, 6]. Plaques usually occur at sites of low shear and nonlaminar turbulent flow such as in the carotid bulb and the proximal internal carotid artery [6], and are rare in the distal part of common carotid arteries where IMT is measured. Both autopsy studies and ultrasonographic studies have demonstrated that carotid plaque is more strongly correlated with atherosclerosis than IMT [7-10]. However, the presence of plaque lacks reliability in detection of atherosclerosis progression, because longitudinally plaque growth along the carotid axis of flow is faster than thickening toward the lumen [11].
We hypothesize that another imaging surrogate such as the sum of all plaque areas in the carotid arteries (total plaque area, TPA) may be more appropriate for the assessment of atherosclerosis progression in observational studies in RA cohorts. TPA represents a carotid plaque burden and has been found to be a stronger predictor of coronary artery disease than IMT [12, 13]. Measuring plaque on a continuous scale increases the ability to quantify the effect of and interaction among risk factors compared with categorical classification (plaque presence). To the best of our knowledge, data on predictive value of TPA in RA cohorts, as well as on progression of TPA and IMT in the same individuals have not been published previously.
Since the morphological components of vessel damage, detected by ultrasound, may reflect different phenotypes of atherosclerosis with distinctive relations to CV risk factors and to clinical vascular disease [5, 14], we have used a complex approach with the assessment of the total carotid plaque burden, pulse wave velocity and local wall elasticity, and arterial hypertrophy by intima-media thickness, adjusting for confounders such as traditional and non-traditional risk factors of CV disease in RA. The main objective was to determine risk factors for TPA at baseline as well as risk factors for carotid plaque progression (∆ TPA) in a small well-characterized group of active RA patients followed serially.
MATERIAL AND METHODS
The study was conducted in full conformance with the principles of the “Declaration of Helsinki” and the protocol was approved by the Health Sciences Research Ethics Committee of the Western University, Ontario, London, Canada. All participants signed an informed consent form, after adequate explanation of the aims, methods, anticipated benefits, and potential hazards of the study.
Study Design
This was an observational single-centre study to identify the CV risk factors (both traditional and non-traditional) that may predict the presence of carotid plaque at baseline and progression over 24 weeks (which was chosen as that is the length of usual RCTs of active RA patients) in order to estimate rates of progression in patients with active RA after adjusting to risk factors.
Patients
The study population was recruited over a period of 1 year and included 31 RA patients (out of 195 screened) with evidence of active disease despite standard of care treatment. Inclusion criteria were age 18-80 years; RA > 3 months’ duration diagnosed according to ACR 1987 criteria; oral corticosteroids (≤10 mg/day prednisone or equivalent) and NSAIDs (up to the maximum recommended dose) if the dose had been stable for at least 2 weeks prior to baseline; moderate or high disease activity - the sum of the swollen joint count (SJC) (66 joint count) plus tender joint count (TJC) (68 joint count) ≥8 at screening OR the sum of the SJC plus TJC ≥4 with elevated acute phase reactants (either hs-CRP≥10 mg/l or ESR≥28 mm/h) at screening or within past 12 months.
To avoid confounding by other inflammatory conditions and treatments, the following exclusion criteria were employed: major surgery within 8 weeks prior to screening with residual surgery related inflammation; rheumatic disease other than RA or uncontrolled significant systemic involvement secondary to RA (e.g. Felty’s syndrome), high doses of steroids (>10 mg/day prednisone or equivalent, including parenteral and intra-articular administration within 4 weeks prior to baseline) and those initiating treatment with cell-depleting therapies within the past six months or/and receiving lipid-lowering medications within 12 weeks of baseline. Participants were excluded if they were pregnant, breastfeeding, had uncontrolled comorbidities, or documented ischemic heart disease (previous infarction, revascularization surgery, angina, or heart failure), cerebrovascular disease (stroke or transient ischemic attack) or symptomatic peripheral artery disease.
Clinical Assessment
Clinical information, including age, sex, and disease duration, history of hypertension, medications, lifestyle (smoking, alcohol, and exercise) was obtained. Height, body weight, waist circumference, and blood pressure were measured. Participants were classified as overweight and obese if BMI was 25-29.9 kg/m2 and ≥30 Kg/m2, respectively. An assessment of 66 joints for swelling and 68 joints for tenderness was made. The 4 variable disease activity score (DAS28) was calculated using erythrocyte sedimentation rate and hsCRP [15, 16]. 100 mm horizontal visual analog scales were used for global assessments of disease activity by the patient and physician, and pain. The Framingham risk score for cardiovascular disease over 10 years was calculated [17]. Clinical characteristics were assessed on the same day as the carotid ultrasound was performed.
Radiographic Assessment
A single radiograph of the hands and wrists was utilized to obtain a radiographic damage assessment using a standardized tool: the Short Erosion Scale (SES) [18]. This scale uses a 6 joint, 12-item scale composed of wrist areas 1,2, and 4 and MCP joints 2,3 and 5 as defined in the Larsen method. A total maximum SES is 60 (12 joints with a maximum score of 5 per joint).
Carotid Ultrasound Measurements
Carotid arteries of RA patients were scanned using a high resolution 3D ultrasound (REF Type of scanner) and the following ultrasound parameters were recorded: presence or absence of plaque, total plaque area (TPA), carotid vessel wall volume (VWV), and Intima-Medial Thickness (IMT). Detailed description of the ultrasound methods used in the study is given in the Supplement 1. TPA was the sum of all plaques between the clavicle and the angle of the jaw in both left and right carotid arteries. Significant plaque burden was determined by a measurement of total plaque area ≥ 0.25 cm2.
Total body composition parameters were assessed by DXA using a Hologic, Model #QDR4500 (place) with standard software supplied by a software 12.3 version. Cockcroft-Gault equation: creatinine clearance was calculated as (140-age) x weight in kg/(72xserum creatinine) in men, and (140-age) x weight in kg/(72 x serum creatinine X 0.85) in women. The laboratory analyses were performed at the time of the carotid ultrasound and included standard hematology and biochemistry assessments, fasting lipid profile (total cholesterol, high-density lipoproteins (HDL), low-density lipoproteins (LDL), triglycerides), hsCRP, ESR, HbA1c, TSH, T4, FSH.
Statistical Analysis
Values were expressed as the mean (standard error; SE) or percentages, as appropriate. Student’s t-test or Mann Whitney U-test was applied for comparison between continuous variables, as well as chi-square or Fisher’s exact test for categorical variables. Spearman’s or Pearson’s tests were used to determine the correlation between TPA, VWV, IMT, PWV, RA parameters and risk factors for atherosclerosis. A stepwise binary logistic regression analysis was performed to determine the factors influencing the presence of plaque. Variables entered into the model were chosen if significantly associated in simple correlation analyses, and those variables known or previously associated with atherosclerosis in RA, from published observations. To identify specific determinants of the plaque progression in RA, linear regression analysis was used. The change in TPA was entered as the dependent variable, and demographic, CVD risk factors and inflammatory variables were considered as independent variables. The results from the multiple stepwise linear regression technique were presented as non-standardized regression coefficients (B) with standard error, P-value and 95% confidence interval. All data were analyzed using SPSS 22.0 software, considering a two-tailed level of P < 0.05 statistically significant.
RESULTS
One hundred ninety five patients with RA were screened, of whom one hundred sixty four were excluded for the following reasons: 64 patients (39%) did not meet the inclusion criterion of disease activity, 39 (24%) received lipid-lowering drugs, 22 (13%) had either malignancy (7 patients) or uncontrolled co-morbidities (15 patients), 3 (2%) underwent recent surgery, 4 (2%) had overlap with other rheumatic diseases, 12 (7%) were older than 80 years, 9 (5%) met exclusion criteria of concurrent therapy (i.e. >10 mg/day prednisone), 11 (8%) were not compliant.
| Clinical characteristics | RA patients with plaque, n = 11 | RA patients without plaque, n = 20 | P-value |
|---|---|---|---|
| Age, years (SE) | 63.2 (8.9) | 57.1 (9.8) | 0.1 |
| Disease duration, years | 18.1 (12.0) | 13.8 (9.5) | 0.29 |
| No. of pts with disease duration ≥10 years, n (%) | 8 (73) | 12 (60) | 0.62 |
| Physician’s global assessment of disease activity (0 to 100 mm) (SE) | 29.1 (18.1) | 20.9 (11.3) | 0.34 |
| Patient’s global assessment of disease activity (0 to 100 mm) (SE) | 38.3 (32.3) | 43.2 (24.1) | 0.40 |
| Pain (0 to 100 mm) (SE) | 32.7 (27.6) | 38.8 (21.1) | 0.28 |
| Short erosion score (SE) | 21.4 (18.5) | 14.6 (15.8) | 0.34 |
| Tender Joint Count (TJC) (0-68) (SE) | 18.8 (10.5) | 18.2 (13.6) | 0.54 |
| Tender Joint Count (0-28) (SE) | 8.6 (5.0) | 9.5 (6.9) | 0.66 |
| Swollen Joint Count (0-66) (SE) | 9.2 (5.3) | 10.0 (4.9) | 0.73 |
| Swollen Joint Count (0-28) (SE) | 7.7 (4.3) | 8.3 (4.1) | 0.99 |
| DAS 28-ESR (SE) | 4.9 (1.1) | 4.8 (1.3) | 0.81 |
| DAS 28-CRP (SE) | 4.5 (1.2) | 4.6 (1.2) | 0.86 |
| Hypertension, n (%) | 7 (64) | 10 (50) | 0.46 |
| Body mass index, kg/m2(SE) | 27.6 (7.8) | 30.6 (5.8) | 0.36 |
| Smoking history: Current smokers, n (%) Former smokers, n (%) Never smokers, n (%) |
9(36) 4 (36) 3 (28) |
6(30) 7(35) 7 (35) |
0.53 |
| Average number of cigarettes/day in smokers (ever) | 12.6 (7.9) | 9.9 (7.6) | 0.19 |
| Number of smoking years (SE) | 20.7 (20.2) | 15.3 (14.6) | 0.40 |
| Pack-years | 19.5 (16.9) | 14.8 (10.4) | 0.32 |
| Family history of cardiovascular disease, n (%) | 5 (46) | 5 (25) | 0.24 |
| Framingham risk score | 19.2 (12.1) | 10.5 (6.6) | 0.005 |
| Creatinine clearance (Cockcroft-Gault equation) | 0.93 (0.36) | 1.16 (0.19) | 0.002 |
| Laboratory parameters | |||
| ESR, mm/hr (SE) | 30.2 (14.2) | 17.7 (15.4) | 0.009 |
| hsCRP, mg/l (SE) | 9.5 (7.7) | 6.7 (6.5) | 0.33 |
| Total cholesterol, mmol/l (SE) | 4.9 (0.8) | 5.1 (0.8) | 0.64 |
| LDL cholesterol, mmol/l (SE) | 2.9 (0.6) | 3.0 (0.7) | 0.42 |
| HDL cholesterol, mmol/l (SE) | 1.52 (0.5) | 1.4 (0.5) | 0.52 |
| Total/HDL cholesterol (AI) (SE) | 3.6 (1.5) | 4.1 (1.5) | 0.42 |
Thirty one patients met inclusion criteria (27 female and 4 male, mean age 59.3±9.8 years). They had median disease duration of fourteen years and a mean DAS28-ESR of 4.8 ± 1.2; where 43% had DAS28 ≥ 5.1. The median SES was 21 (range from 0 to 60). Ninety-seven percent were receiving disease modifying anti-rheumatic drugs (DMARDs) at screening visit, alone or in combination: oral hydroxychloroquine (n=8), weekly oral methotrexate or injectable methotrexate (n= 18), leflunomide (n=7), sulfasalazine (n=8), D-penicillamine (n=1), and TNFα inhibitors (n=10). Eleven patients were taking daily oral prednisone (n=11) at baseline.
Cardiovascular Risk Factors in Patients with RA
Traditional CV risk factors were frequent among the RA patients, including hypertension (55%), smoking (84%), physical inactivity (61%), obesity (45%), dyslipidemia (68%), diabetes mellitus (7%), and family history of cardiovascular disease (32%).
At study entry, we identified 11 subjects with high atherosclerotic burden defined as a measurement of total plaque area (TPA) ≥ 0.25 cm2 (Table 1). Those patients had a significantly greater Framingham risk scores (14.7±9.65 vs. 8.05±3.8, p=0.005) and reduced glomerular filtration rate (0.93±0.36 vs. 1.16±0.19, p=0.002), an independent risk factor for cardiovascular disease.
From the disease-related variables, ESR was significantly higher in RA patients with TPA ≥ 0.25 cm2 than in those without plaque burden (30.18±14.21 vs. 17.68±15.37, p=0.009); all other parameters of disease activity and/or severity were comparable in both groups. There was no difference in atherosclerotic burden for disease duration, DAS 28, and SES.
Stepwise binary logistic regression analyses showed that ESR (OR=1.148, 95%CI: 1.015-1.299, p=0.028) and Framingham Risk Factor Score (OR=1.155, 95%CI: 1.002-1.332, p=0.046) were significant predictors of plaque. Traditional risk factors (Framingham risk score) predicted the high atherosclerotic burden in 70%. 30% of patients classified by Framingham risk score as low risk for CV events (<10%) had US-measures of significant atherosclerosis. Modified Framingham risk score (1.5 multiplication factor) failed to identify 22% of the patients with high atherosclerotic burden (TPA≥0.25cm2) and 20% of those with maxIMT≥0.8 mm.
The groups of patients with and without high atherosclerotic burden were formed on the basis of TPA measurements, and TPA itself had a significant association with ESR (r=0.49, p=0.006) and Framingham risk score (r=0.527, p=0.008).
Most patients (86%) had increased arterial thickness (maxIMT≥0.6mm) and half of them had maxIMT≥0.8mm. Arterial stiffness and thickening was linked to smoking (pack-years was associated with PWV (r=0.64, p=0.008) and IMT (r=0.48, p=0.04)). PWV showed a positive association with age (r=0.53, p=0.004). Carotid artery vessel wall volume did not show any statistically significant difference between the groups of RA patients with and without significant plaque burden (374.96±56.8mm3 vs. 334.16±71mm3, p=0.12) and it was not associated with RA-related parameters and traditional CV risk factors. Body mineral density (BMD) of lumbar spine was slightly lower in the group of RA patients with plaque (0.97±0.17 b/cm2 vs. 1.06±0.12 b/cm2, p=0.05), but all other parameters (BMD of femur, total body mineral content, total fat, total lean, total mass, percentage of fat) were similar in those with and without high atherosclerotic burden.
Inflammation and Lipid Metabolism in Rheumatoid Arthritis
An atherogenic lipid profile characterized by reduced HDL levels and/or elevated atherogenic index (AI) occurred in 68% of RA patients. The measures of disease activity (global assessments of disease activity by physician and by patient, DAS-28, TJC 68, SJC 66) showed a strong positive association with dyslipidemia (Table 2).
| Lipid blood level | DAS 28-hsCRP | DAS 28-ESR | Disease activity assessed by physician | Disease activity assessed by patient | TJC68 | SJC66 | |
|---|---|---|---|---|---|---|---|
| HDL, mmol/l | R=-0.55, p=0.006 | R=-0.67, p=0.0001 | R=-0.11, p=0.59 | R=-0.23, p=0.27 | R=-0.422, p=0.036 | R=-0.54, p=0.005 | |
| LDL, mmol/l | R=0.24, p=0.3 | R=0.39, p=0.06 | R=0.39, p=0.049 | 0.46, p=0.02 | R=0.4, p=0.047 | R=0.26, p=0.2 | |
| AI | R=0.627, p=0.001 | R=0.79, p=0.0001 | R=0.38, p=0.06 | R=0.49, p=0.01 | R=0.59, p=0.002 | R=0.57, p=0.003 | |
| Total Cholesterol, mmol/l | R=0.045, p=0.83 | R=0.1, p=0.6 |
R=0.42, p=0.028 | R=0.35, p=0.08 | R=0.25, p=0.2 | R=0.12, p=0.55 | |
The hsCRP blood levels were related to disease activity as measured by Physician’s global assessment (r=0.443, p=0.016), and traditional CV risk factors: LDL (r=0.544, p=0.007), family history of CV events (r=0.464, p=0.011), and smoking (pack-years) (r=0.384, p=0.4, n=29).
| Predictor | Unstandardized Coefficients | Standardized Coefficients | t | Sig. | 95.0% Confidence Interval for B | ||
|---|---|---|---|---|---|---|---|
| B | Std. Error | beta | Lower Bound | Upper Bound | |||
| hsCRP, mg/l | .020 | .005 | .844 | 4.451 | .002 | .010 | .030 |
| ESR, mm/h | .013 | .004 | .789 | 3.401 | .011 | .004 | .021 |
| Daily N. of cig. | .025 | .007 | .838 | 3.434 | .019 | .006 | .043 |
| Pack-years | .011 | .003 | .892 | 4.411 | .007 | .005 | .018 |
Predictors of Plaque Progression
Ten patients from the primary cohort who had baseline significant plaque burden were recruited into a 24 Week prospective study. The medications taken for RA during the 24-week period remained stable. One patient was lost to follow-up. All of the assessments described above were conducted at baseline, and repeated at 24 weeks. There were no changes found between baseline and FU IMT measurements (0.763±0.134mm and 0.769±0.133mm, respectively). Mean (±SD) TPA at baseline was 0.43±0.18cm2 and 0.52±0.29cm2- at FU visit. Fig. (1) shows the plaque progression in
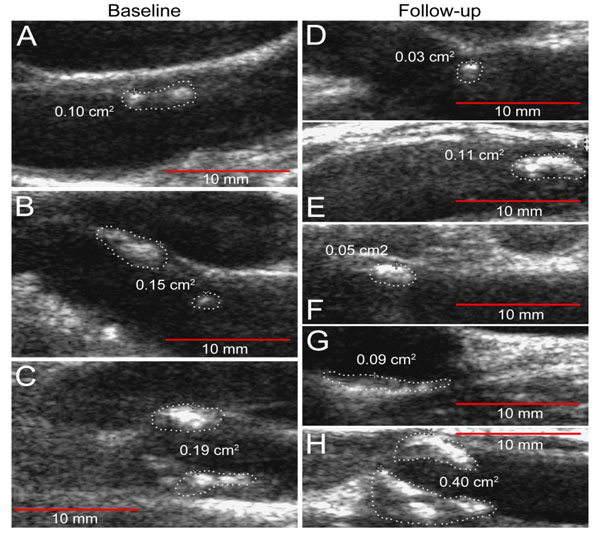
RA patient. ∆TPA did not always parallel ∆IMT: three patients with an increase in TPA measurements (∆TPA 0.1, 0.24 and 0.45cm2) did not show any IMT changes. Linear regression analysis was performed to determine predictors (independent variable) of the plaque progression assessed by the pairwise comparison of baseline TPA and the subsequent measurement time (dependent variable) (Table 3). The ESR and hsCRP were observed to have a significant effect on the increase in plaque size (β=0.79, p=0.01 and β=0.85, p=0.002, respectively), as well as smoking (number of pack-years: β=0.89, p=0.007, and daily number of cigarettes: β=0.84, p=0.02) by univariate linear analysis. Because of the limited number of RA patients who had baseline plaque, we evaluated the relationship between the predictor and outcome controlling for other predictors by introducing into the models (stepwise method) hsCRP together with (one at a time) traditional risk factors and RA-related parameters. Stepwise linear regression analysis revealed that only hsCRP was a significant risk factor for TPA progression in RA patients (B=0.019, SE=0.003, β=0.947, p=0.004). Smoking and ESR were cofounders associated with hsCRP. We did not find any associations between disease characteristics or traditional CV risk factors and ∆IMT by linear regression analysis.
DISCUSSION
We observed a high prevalence of 3DUS measurements of significant atherosclerotic burden (35%) and arterial wall hypertrophy (86%) in RA patients with long disease duration and active RA at baseline. These findings were greater than would be expected in age and sex matched controls [19]. Similar to our findings, Roman et al. (2006) reported that plaque was three times more prevalent in the patients with RA than in controls in all decades of life [20]. Another cross-sectional study also revealed an increased prevalence of severe subclinical atherosclerosis in long standing RA: 72% of RA patients had atherosclerosis and 31% had plaque in that cohort [21].
Traditional risk factors such as hypertension, type 2 diabetes, smoking, hypercholesterolemia, obesity and physical inactivity explain at least some of the excess CV risk in RA patients [22, 23]. We observed that the Framingham risk score as a composite measure of the burden of traditional risk factors predicted atherosclerotic plaque in RA, and smoking was associated in univariate analysis with worsening atherosclerotic plaque at 24 weeks. In recent meta-analysis, Baghdadi et al. showed that CV events in RA patients were strongly associated with hyperlipidemia and hypertension, while RA duration and erosions were less significant contributors [23]. However, the application of traditional CV risk factor assessment equations, such as Framingham, Reynolds risk score and the Systematic Coronary Risk Evaluation models, to patients with RA is reported to underestimate their CV risk [24]. Accordingly, we found that Framingham risk score predicted the high atherosclerotic burden only in 70% of patients.
Systemic inflammation significantly contributes to the rapid progression of subclinical atherosclerosis in RA [25]. We found that hsCRP and ESR were strongly associated with atherosclerotic plaque growth over 24 weeks. Moreover, ESR was the best predictor of atherosclerotic burden in our primary RA cohort. Similar to our results, some other studies have demonstrated a significant association between ESR and the risk of CVD [26, 27]. The CRP at baseline has been found to be an important predictor of subsequent death from CV disease in patients with new onset inflammatory polyarthritis and was independent of other factors of disease severity [28]. The magnitude and chronicity of the inflammatory response, as measured by CRP, correlated with carotid atherosclerosis development in RA [29]. We found that hsCRP levels were significantly linked to LDL in RA. It is also possible that under high inflammatory burden, excessive production of acute phase reactants may impair trafficking of cholesterol in the liver or impede normal cholesterol production. RA-related inflammation not only influences the plaque progression, but also promotes plaque vulnerability. Higher plaque vulnerability has been reported in patients with active RA, potentially contributing to future CV events [30]. These findings point to the critical importance of adequate disease control in lowering CV risk. The modified CV risk evaluation model, which would incorporate the impact of systemic inflammation, has yet to be developed. The use of 1.5 multiplication factor recommended for RA patients [31] did not improve risk estimation in our RA cohort: modified Framingham risk score failed to identify a quarter of patients with significant atherosclerotic burden detectable by 3DUS.
Most patients with active RA had an atherogenic lipid profile: low HDL and thus an increased TC/HDL ratio (atherogenic index), which is an important prognostic marker for CVD [32]. The available literature on lipid profiles in patients with RA is contradictory [33, 34]. A higher disease activity measured by DAS 28 and Physician’s global assessment in our study was associated with higher LDL levels, lower total cholesterol and HDL levels, leading to an unfavorable atherogenic index. Similar to our study, the serum total cholesterol and HDL levels in RA were demonstrated to be inversely linked to disease activity: patients in remission or with controlled disease showed an increase in HDL levels and a reduction in the atherogenic index compared to patients with active disease [35]. These findings have been recently confirmed by a larger cross-sectional study in 204 patients with RA, demonstrating an adverse effect of disease activity on lipid profile [36]. However, the temporal and casual relationships between inflammation and changes in lipids are still unclear. RA-related inflammatory milieu might adversely affect the lipid profile directly via. cytokine production (TNF-α, IL-6), as well indirectly leading to the modifications of lifestyle factors (i.e impaired physical activity) and sharing a common genetic predisposition to RA and dyslipidemia [37-39].
We must acknowledge a number of study limitations. First, this was a small pilot study, involving only subjects who met the strict inclusion and exclusion criteria and consented to US monitoring. Only active RA patients were included which limits the generalizability of findings (most RA patients who were screened were not eligible). This could also affect power to detect the link between disease activity measures and atherosclerosis in a cross-sectional portion of the study, because disease activity range was small across the recruited subjects. In chronic inflammatory diseases such as RA, the individual inflammatory markers’ concentrations (i.e. hsCRP) frequently fluctuate during the course of disease making the impact of such changes on CV risk less clear. To overcome this limitation, we used some surrogate measures of disease progression, such as the ratio of joint damage over disease duration, and the number of DMARDs used in the past, as an attempt to quantify the total inflammatory burden of disease. The burden of inflammation seems to be more important than isolated measurement of inflammatory markers, such as CRP, for progression of atherosclerosis and cardiovascular mobility and mortality. Further longitudinal studies with follow-up assessments of radiographic damage scores over observation period are warranted to estimate a total effect of the chronic inflammation on atherosclerosis progression.
The effect of RA treatment on atherosclerosis is important to consider. The proportion of patients in our study was receiving non-biologic DMARD therapies, and/or TNF-inhibition therapy. TNF-α inhibitors and methotrexate may be cardioprotective in RA [40] and thus influence the study results. For tracking atherosclerotic lesion changes over time, we used a 3DU carotid ultrasound measurement of TPA. Our approach to the evaluation of the carotid ultrasound parameters was based on a recent randomized placebo controlled study [41], where the treatment with high dose atorvastatin resulted in a statistically significant and clinically relevant decrease in TPA. These measurements have also been utilized in a diabetic patient population providing greater dynamic range and allowing for statistically significant differences in plaque progression as compared to IMT progression [42-44]. IMT is a small structure, and changes over time representing only tenths of millimeter, that is, the resolution of the B-mode image is below the quantities being measured. Indeed, we did not find any reliable associations of IMT changes over time with CV risk factors in our group of patients. The fact that plaques are larger structures makes TPA more suitable for longitudinal measurements at an individual level.
CONCLUSION
In conclusion, a high prevalence of carotid artery atherosclerosis was detected in active RA with long disease duration. Along with the increased burden of traditional cardiovascular risk factors, RA-related systemic inflammation significantly contributed to the occurrence and progression of atheroma and might mediate its effects on atherosclerosis through hsCRP and modulation of traditional risk factors, such as dyslipidemia. Thus, achieving disease remission is crucially important in lowering CV risk in RA patients; a new CV risk estimation model or adjustment method in RA patients is required.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

