All published articles of this journal are available on ScienceDirect.

IgG4-Related Lung Disease Associated with Usual Interstitial Pneumonia
Authors Info & Affiliations
Abstract
We report a case of immunoglobulin(Ig)G4-related disease with the radiologic and histopathological manifestations resembling usual interstitial pneumonia (UIP). The patient was a 62-year-old man who presented with progressive dyspnea of insidious onset. High resolution computed tomography of the chest showed lower-lobe predominant peripheral reticulation and traction bronchiectasis but no honeycomb change. Microscopic examination of the surgical lung biopsy showed characteristic features of UIP including architectural distortion by fibrosis with peripheral and paraseptal accentuation, scattered fibroblast foci and microscopic honeycomb change. In addition there were prominent multifocal lymphoplasmacytic infiltrates with a marked increase of IgG4-positive plasma cells (79 per high power field in hot spots) and high IgG4/IgG ratio (up to 67%). The serum IgG4 level was elevated at 760 mg/dl (reference range 9-89), with normal levels for the other IgG subclasses and negative serologic markers for autoimmune diseases. The patient’s symptoms improved significantly with oral corticosteroid treatment.
INTRODUCTION
Immunoglobulin(Ig)G4 -related disease (IgG4-RD) is a chronic fibroinflammatory condition characterized by tumefactive/infiltrative fibrosis and lymphoplasmacytic infiltrates involving a single or several anatomic sites and often with an elevated IgG4 serum level [1]. Awareness of this entity has increased significantly over the last decade, but its cause and pathogenesis remain incompletely understood [2]. Autoimmune pancreatitis is the originally described organ manifestation of IgG4-RD [3]. Many other anatomic sites have been reported since, including salivary and lacrimal glands, lymph nodes, spleen, lung, pleura, breast, liver, bile ducts, gallbladder, retroperitoneum, kidney, prostate, paravertebral space, mediastinum, meninges, peripheral nerves and arteries [4].
A recent consensus statement suggests using the following major histopathologic features to make a diagnosis of IgG4-RD: (a) dense lymphoplasmacytic infiltrate, (b) fibrosis, arranged at least focally in a storiform pattern, and (c) obliterative phlebitis [5]. Although the serum IgG4 level is often elevated in IgG4-RD, it can be normal in up to 40% of patients [6]. Compared with other sites, pulmonary involvement by IgG4-RD is often difficult to recognize due to a relative paucity of pathognomonic features and a plethora of overlapping findings with other fibroinflammatory diseases [22, 23]. The lung is one of the sites in which storiform fibrosis and obliterative phlebitis can be minimal or absent [7]. Lung involvement by IgG4-RD often shows lymphoplasmacytic and eosinophilic inflammation and fibrosis involving interlobular septa, peribronchiolar spaces and pleura [8]. Here we describe a case of IgG4-RD with radiologic and pathologic features of UIP, a rarely described manifestation.
Report of a case
A 62-year-old man with a past medical history notable only for coronary artery bypass grafting nine years prior presented with insidious onset of dry cough and dyspnea for approximately six months. He denied sputum production, hemoptysis and pleuritic chest pain. He had no SICCA symptoms, arthralgias, myalgias, muscle weakness, Raynaud phenomenon, or mechanic's hands. He reported a 10 year history of pigment loss over his face, bilateral hands and inguinal regions but denied any other skin changes or rashes. There was no suggestion of sleep apnea or aspiration. He was a never-smoker and worked in an office setting without any apparent environmental, occupational or pneumotoxic medication exposures.
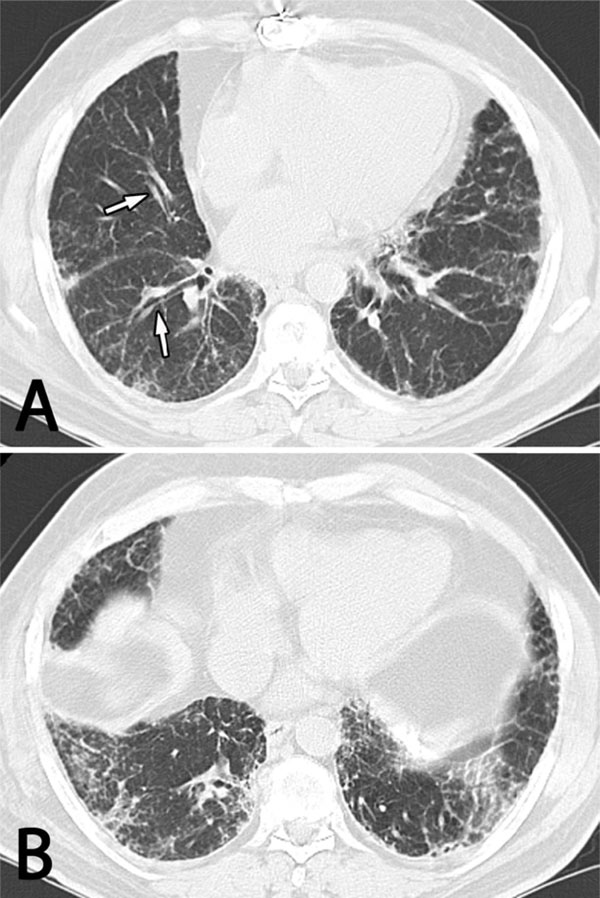
High resolution computed tomography (HRCT) of the chest showing diffuse bilateral interstitial abnormalities with subpleural accentuation that are more prominent in the lower (B) than the mid (A) and upper (not shown) lung zones. There is thickening of the bronchovascular bundles with mild bronchiectasis (arrows) but no honeycomb change.
Physical examination was notable for bi-basilar mid-inspiratory crackles and hypopigmentation in a distribution consistent with vitiligo, raising the question of localized scleroderma, but his history and physical exam were not otherwise suggestive of underlying autoimmune diseases.
Serologic testing for SS-A, SS-B, anti nuclear antibodies (ANA), anti-neutrophil cytoplasmic antibodies (ANCA), rheumatoid factor, (RF) aldolase, anti-topoisomerase I (Scl-70) antibodies and anti-citrullinated peptide (CCP) antibodies was negative. His thyroid stimulating hormone (TSH), erythrocyte sedimentation rate and C-reactive protein level were normal. Complement component C3 was in the mid normal range while C4 was low normal. Mild peripheral eosinophilia of 10% was noted with an absolute eosinophil count of 0.60 x 109/L (reference ranges 0-6 and 0.00-0.40x109, respectively) and IgE was increased to 609 IU/mL (reference range < 160). Serum protein electrophoresis (SPEP) revealed mild elevation of the gamma globulin fraction of 1.85 gm/dl (reference range 0.70-1.60). No monoclonal proteins were detected.
Pulmonary function tests (PFTs) showed a mild restrictive pattern with a moderately decreased diffusing capacity for carbon monoxide (DLCO). High resolution computed tomography (HRCT) of the chest showed diffuse bilateral non-specific interstitial abnormalities with lower zone predominance (Fig. 1). There was mild traction bronchiectasis at the bases and mid-zones but no honeycomb change. The mid-esophagus was mildly dilated. A calcified pleural plaque was present on the diaphragmatic surface, presumably related to a hemothorax that had complicated his prior cardiac surgery.
The patient’s case was discussed at our multidisciplinary autoimmune lung disease conference, and the consensus opinion was to proceed with surgical lung biopsy. Microscopic examination of wedge biopsies obtained from all three right lung lobes showed a chronic fibrosing lung disease with peripheral and subpleural accentuation, fibroblastic foci and microscopic honeycombing with inspissated mucus (Fig. 2). Histopathologic features met diagnostic criteria for UIP [9]. There were increased number of lymphoid aggregates with germinal centers as well as a prominent lympho-plasmacytic infiltrates, mainly involving thickened pleura and interlobular septa, prompting immunohistochemical stains for IgG4 and IgG. The IgG4 stain showed 79 IgG4-positive plasma cells averaged over the three high power fields in hot spots Fig. (3), with an IgG4/IgG ratio of 67%. No obliterative phlebitis or arteritis was found. Plasma cells were polytypic by immunohistochemical stains for immunoglobulin kappa and lambda light chains.
In light of high IgG4-positive cell counts, a serum IgG4 level was found to be markedly elevated at 760 mg/dl (reference range 9-89). Other IgG subclass levels were normal. The diagnosis of IgG4-RD was reached by multidisciplinary consensus, and the patient was treated with 20 mg of prednisone twice a day. There was no overt involvement of other organ systems by IgG4-RD, but we did not screen exhaustively for subclinical or asymptomatic organ involvement.
Within two weeks of starting systemic corticosteroids the patient reported a significant subjective improvement in his dyspnea and fatigue with only mild residual cough. Unfortunately, three months after his initial presentation the patient died of acute respiratory failure, presumably due to a lower respiratory tract infection based on elevated white blood cell count and radiographic consolidation with air bronchograms. An acute exacerbation of the underlying lung disease could not be excluded since bronchoalveolar lavage studies failed to identify an infectious agent. A post-mortem examination was not performed.

Usual interstitial pneumonia (UIP) as manifestation of IgG4-related disease. Low power (A and B) and high power (C and D) views of the surgical lung biopsy show typical distribution of fibrosis along pleura and interlobular septa with spared centrilobular areas and fibroblastic foci (arrows). Superimposed is a prominent lymphoplasmacytic inflammatory infiltrate involving pleura and interstitium (arrowheads). [Hematoxylin Eosin, original x20 (A and B) and x200 (C and D)]
DISCUSSION
We report a case of IgG4-RD mimicking idiopathic pulmonary fibrosis (IPF) clinically, radiologically, and histopathologically. Histopathologic features of UIP were accompanied by a prominent IgG4-positive plasma cell infiltrate, a feature atypical for IPF [10,11]. While we have encountered one similar case (TVC, ESY unpublished observation), IgG4-RD presenting as fibrosing interstitial lung disease with a UIP pattern is unusual in our experience. Conversely, increased IgG4-positive cells are not seen in most UIP cases [10].
Radiographically, the majority of the approximately 100 cases of IgG4-related lung disease described in the literature to date show one of the following patterns: (a) mass lesions, (b) patchy consolidation, or (c) interstitial lung disease [12].
Histopathologically, IgG4-RD presents as nodular, bronchovascular or alveolar septal/interstitial disease with varying degrees of pleural involvement [7]. It is characterized by dense lymphoplasmacytic infiltrates containing IgG4-expressing plasma cells, storiform fibrosis and obliterative phlebitis [5]. Interstitial disease often resembles non-specific interstitial pneumonia (NSIP) [21]. Histopathologic features usually, but not always, correlate with the radiographic impression. For example, Yamashita et al. found histopathologic involvement of bronchovascular bundles in patients who presented with nodular disease radiographically [13, 14].
Two coexisting processes remain difficult to exclude, particularly because the terminal event might be interpreted as an acute exacerbation of his ILD. Acute exacerbations are well recognized in the setting of IPF, but have also been described among other fibrosing interstitial pneumonias. Although this patient responded well to systemic corticosteroid therapy, the long term prognosis of UIP in the setting of IgG4-RD is largely unknown, especially when compared with idiopathic or connective tissue disease-associated UIP.
Diagnostic criteria for IgG4-RD were proposed in a recent consensus statement [5]. The absolute numbers of IgG4-positive plasma cells and the IgG4-to-IgG ratio needed to support a diagnosis of IgG4-RD depends on the anatomic site and on how many other histologic features are present to support the diagnosis. Our case showed a dense lymphoplasmacytic infiltrate that met both proposed thresholds: more than 50 IgG4-positive plasma cells per high power field and an IgG4/IgG ratio of greater than 40%. Although fibrosis was abundant, it was not particularly storiform in any area, nor were the veins involved by obliterative phlebitis. This is in keeping with Zen’s observation of obliterative phlebitis in 90% of their patients with nodular and pleural, but only 14% of those with interstitial or bronchovascular involvement [7]. Similarly, storiform fibrosis appears to be more common in tumefactive or pleural lesions as opposed to cases with mostly alveolar septal involvement [7]. The same group identified lymphoid follicles in half of their patients as was seen in our case.
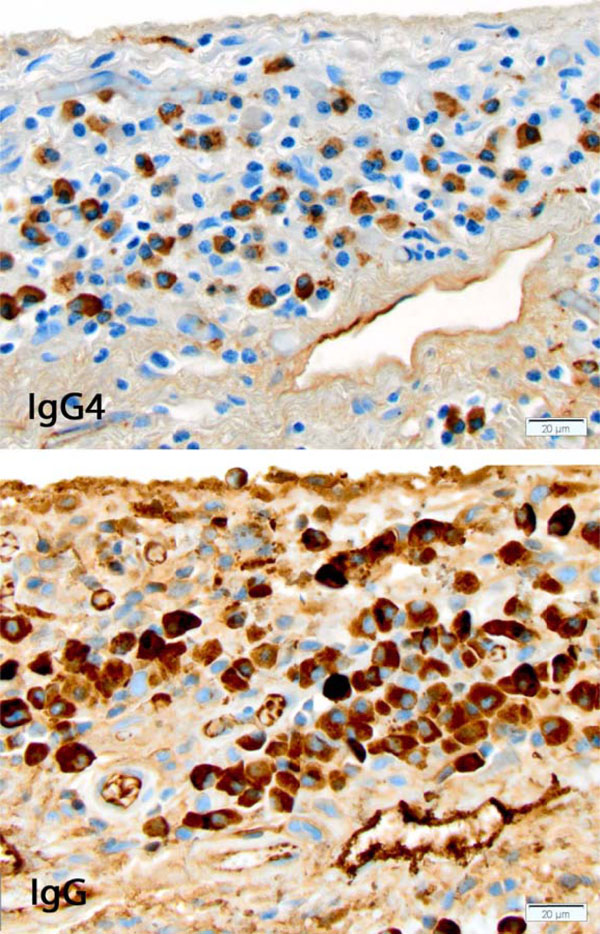
IgG4-related disease involving pleura. Portion of a high power field showing the dense lymphoplasmacytic infiltrate with numerous plasma cells, many of them expressing IgG and IgG4. [Immunohistochemical stains for IgG4 (top) and IgG (bottom), original x400]
Extrathoracic lesions seem to be common in patients with lung involvement by IgG4-RD and may help suggest or ascertain the diagnosis. Matsui et al. noted them in all of their 18 patients and Zen et al. in 9 of their 21 patients (43%) [7, 8]. Interestingly, our patient had experienced mild pigment loss for about 10 years involving hands, face and inguinal regions. Although not a known association with IgG4-RD, a recently reported cohort of 60 patients included one patient with vitiligo [15]. Since autoantibodies of IgG4 subclass have been described in Wegener granulomatosis (granulomatosis with polyangiitis, or GPA) and various other autoimmune diseases, one could hypothesize that among the IgG4 antibodies produced in our patient was an anti-melanocyte antibody that resulted in vitiligo [16]. One of the authors has encountered a patient with IgG4-RD who developed an ANCA that resolved with rituximab-induced remission of the IgG4-RD (MCL, unpublished observation).
UIP occurring in the setting of connective tissue disease is often characterized by prominent or even exuberant interstitial inflammation and lymphoid hyperplasia with germinal centers [17]. Distinguishing IgG4-RD presenting as ILD from the more common connective tissue diseases could be challenging since serologic testing in IgG4-related ILD may reveal antibodies associated with rheumatologic diseases. Takato’s NSIP patient, for example, had anti-nuclear antibodies showing a homogenous immunofluorescence pattern [18]. Two of 18 patients in Matsui’s series were found to have Sjögren disease-associated antibodies SS-A/SS-B [8]. Ahn also found SS-A antibodies in a patient they believe most likely had IgG4-RD although the density of IgG4-positive plasma cells did not meet the proposed diagnostic criteria [24]. Jinnur’s patient with cavitating lung disease had anti-nuclear antibodies [19]. Distinguishing IgG4-RD from Sjögren disease can be especially difficult, an overlap highlighted by the fact that Mikulicz syndrome, an inflammatory condition of salivary glands historically considered a subgroup of Sjögren disease, is currently thought of as a manifestation of IgG4-related disease [20]. On the other hand, Shrestha et al. reported that none of their patients with Sjögren disease had increased IgG4-positive plasma cells in the lung biopsies [10].
In summary, UIP with heavy lymphoplasmacytic infiltrates can be a manifestation of IgG4-RD. The study of additional cases of UIP in the setting of IgG4-RD would improve our understanding of the natural history and therapeutic approach to this subtype.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGEMENTS
This study was funded with departmental funds of the University of Pittsburgh Medical Center Department of Pathology. There were no sponsors associated with this report: the design; data collection, analysis, and interpretation; preparation, review of the manuscript and decision to submit the manuscript for publication were all completed by the authors.

