All published articles of this journal are available on ScienceDirect.

Two-Year Safety and Efficacy Experience in Patients with Methotrexate-Resistant Active Rheumatoid Arthritis Treated with Etanercept and Conventional Disease-Modifying Anti-rheumatic Drugs in the Latin American Region
Authors Info & Affiliations
Abstract
Background:
Although long-term data are available from biologic studies in North American/European populations with rheumatoid arthritis (RA), long-term findings in Latin American RA populations are limited.
Objective:
To examine long-term safety/efficacy of etanercept, methotrexate, and/or other disease-modifying anti-rheumatic drugs (DMARDs) in Latin American patients with moderate-to-severe active RA.
Methods:
In the first phase of this open-label study, patients were randomized to etanercept 50 mg weekly plus methotrexate or conventional DMARD (hydroxychloroquine or sulfasalazine) plus methotrexate for 24 weeks. At the start of the second phase (week 24), investigators selected a treatment regimen that included any combination/dosage of etanercept, methotrexate, hydroxychloroquine, or sulfasalazine based on previous treatment response, preference, and local product labeling, and was continued for the 104-week extension.
Results:
In the extension, in the group previously randomized to etanercept-plus-methotrexate therapy, etanercept was continued in 259/260 patients; methotrexate continued in 260/260; and hydroxychloroquine and sulfasalazine added in 8/260 and 3/260, respectively. In the group previously randomized to conventional DMARD-plus-methotrexate therapy, conventional DMARD was discontinued in 86/126 and etanercept added in 105/126. Among etanercept-exposed patients (total exposure, 798.1 patient-year [PY]), rates of adverse events, serious adverse events, and serious infections per PY were 1.7, 0.07, and 0.02 events per PY. In both groups, after treatment modification was permitted, clinical response rates and improvements in clinical/patient-reported outcomes from baseline were sustained to week 128.
Conclusion:
After investigators were permitted to modify treatment, etanercept was part of the treatment regimen in 95% of patients. Continuation or addition of etanercept in the 2-year extension resulted in a consistently good risk:benefit profile.
Trial Registration:
Open-Label Study Comparing Etanercept to Conventional Disease Modifying Antirheumatic Drug (DMARD) Therapy; ClinicalTrials.gov, number NCT00848354; https://clinicaltrials.gov/ct2/show/NCT00848354
INTRODUCTION
Clinical trials in rheumatoid arthritis (RA) commonly include patients from many countries, but relatively few have included patients from Latin America, where the prevalence of this chronic, potentially crippling and progressive condition is estimated to be 0.4% [1-3]. Latin America is distinct from North America and Europe, the regions in which most RA clinical trials have been conducted, in a number of important ways [1, 2]. The population of Latin America is racially and ethnically diverse, including indigenous peoples genetically distinct from Western populations [4]. Several barriers to prompt diagnosis and treatment of RA remain in this region, including the distance to or lack of local specialist clinicians and a low public awareness of the disease. In addition, public health efforts in Latin America have traditionally been focused on conditions that substantially increase mortality such as infectious diseases and maternal/child care; although RA is associated with increased morbidity and mortality, such chronic disabling diseases largely have been overlooked [2]. Currently, rheumatology societies in Latin America are making major advances in educating clinicians about RA, but region-/country-specific information remains sparse. Remission of RA is increasingly recognized as a realistic treatment goal, primarily because of the effective use of methotrexate, other synthetic disease-modifying anti-rheumatic drugs (DMARDs), and biologics such as anti-tumor necrosis factor (TNF) agents [5-8]. According to treatment recommendations of the Pan American League of Associations for Rheumatology (PANLAR) and Grupo Latino Americano de Estudio de Artritis Reumatoide (GLADAR), patients diagnosed with RA should receive at least 3 months of conventional DMARDs, including methotrexate, hydroxychloroquine, and sulfasalazine, per local standards of care before initiating treatment with a biologic [9]. Biologic agents are reserved for patients who continue to experience joint damage and pain despite standard-of-care DMARD therapy or who have intolerable adverse effects related to DMARDs [9]. In addition to the PANLAR/GLADAR recommendations, clinicians in Latin America need to consider individual country and regional guidelines and local product labeling. The efficacy and safety of the anti-TNF agent etanercept, administered in combination with methotrexate or as monotherapy, have been well established in clinical studies of moderate-to-severe RA [10, 11]. Although patients from Latin American countries were included in these studies, subset populations were too small to allow comparative analyses with global populations. In an initial report of 24-week findings of a two-phase, randomized, open-label study conducted in the Latin American region (ClinicalTrials.gov, number NCT00848354), treatment with the combination of etanercept plus methotrexate was compared with the conventional DMARDs hydroxychloroquine or sulfasalazine plus methotrexate, as selected and dosed by the investigator to reflect standard of care and the local label, in patients with active RA who were experiencing a suboptimal response to methotrexate monotherapy [12]. Here, we present extended data on safety and clinical efficacy and patient-reported outcomes (PROs) over an optional 104 weeks of follow-up care after investigators were permitted to modify the initial randomized treatment regimens.
METHODS
Study Design and Patients
A detailed description of the first 24-week study phase, initiated in June 2009 in five countries in Latin America (i.e. Argentina, Chile, Colombia, Mexico, and Panama), has been published elsewhere [12]. In brief, adult RA patients who had active disease (≥8 tender/≥6 swollen joints and erythrocyte sedimentation rate [ESR] ≥28 mm/h) despite treatment with methotrexate 7.5 to 25 mg/week for at least 3 months were randomized in a 2:1 ratio to receive etanercept (50 mg/week) plus methotrexate or the conventional DMARD hydroxychloroquine or sulfasalazine (at the investigator’s discretion) plus methotrexate for 24 weeks. Methotrexate doses could be titrated between baseline and week 4, from a minimum dosage of 7.5 mg/week to a maximum dosage of 25 mg/week, but stable doses were required thereafter. In patients with treatment-related adverse events, methotrexate doses could be reduced, with doses re-titrated to previous levels when possible. Hydroxychloroquine and sulfasalazine were titrated to optimal doses by week 4 based on investigator judgment to reflect local standard of care and product labeling. Permitted concomitant medications included stable doses of oral corticosteroids (≤10 mg/d prednisone or equivalent), intra-articular corticosteroid injections (administered >28 days prior to study visits), and a single non-steroidal anti-inflammatory drug (as-needed, with dose adjustments and substitution allowed).
Patients who completed the 24-week randomized phase could enter an open-label, 104-week extension phase (for a total of 128 weeks of treatment). At the start of the optional study extension, investigators selected a treatment regimen (i.e. etanercept, methotrexate, hydroxychloroquine, or sulfasalazine in any combination at the desired dosage) based on patients’ previous response to randomized treatment, their preference, and local product labeling. Biologic agents other than etanercept or conventional DMARDs other than hydroxychloroquine, sulfasalazine, or methotrexate could not be used. Doses of methotrexate, hydroxychloroquine, or sulfasalazine selected at week 24 could be titrated at the discretion of the investigator, but no new treatment could be initiated after treatment selection for patients entering the extension. Although the selected treatment was to remain as stable as possible throughout the subsequent 104 weeks of follow-up, the investigator could make therapeutic changes based on adverse events (i.e. titration or discontinuation of a selected treatment or substitution with an alternate study DMARD) or disease activity (titration or discontinuation of a selected treatment).
This study was conducted in compliance with the Declaration of Helsinki principles, the International Conference on Harmonisation Good Clinical Practice guidelines, and local regulatory requirements. All patients signed informed consent forms, which were reviewed and approved by independent ethics committees or institutional review boards.
Assessments
Changes in the initial randomized treatment regimens were recorded at the beginning of the optional extension. Treatment-emergent adverse events and serious adverse events were assessed at each study visit and classified by system organ class and preferred terms of the Medical Dictionary for Regulatory Activities. Clinical efficacy end points in the extension included the proportions of patients achieving the American College of Rheumatology (ACR) 20 (ACR20), 50 (ACR50), and 70 (ACR70) responses (20%, 50%, and 70% improvement in ACR response criteria); low disease activity (LDA), defined as a Disease Activity Score in 28 joints (DAS28) based on ESR of <3.2; and remission, defined as DAS28 <2.6. Mean changes from baseline to weeks 76 and 128 were assessed for DAS28; tender and swollen joint counts; physician global assessment; and duration of morning stiffness.
PROs were evaluated using the subject global assessment; visual analog scales for general health, pain, and fatigue; the total Health Assessment Questionnaire Disability Index (HAQ-DI); the physical and mental component summaries (PCS, MCS) and vitality domain of the 36-item Short-Form (SF-36) Health Survey; and the Work Productivity and Activity Impairment:RA (WPAI:RA) questionnaire. The proportions of patients achieving a normal score on the HAQ-DI (i.e. ≤0.5) and a minimally clinically important difference (MCID; i.e. ≥5) in improvement from baseline in the SF-36 scores for the PCS, MCS, and vitality domain were assessed. Physician and patient satisfaction with RA control were evaluated throughout the 104-week extension using questionnaires. The Caregiver Burden and Resource Utilization (CBRU) questionnaire evaluated current employment and utilization of healthcare resources (i.e. rheumatologist and emergency department visits and caregiver assistance).
Statistical Methods
Results of all analyses from the extension were summarized using descriptive statistics; no statistical comparisons of findings from the 104-week extension were performed. Findings from the extension are summarized using the treatment groups to which patients were randomized in the initial randomized phase (i.e. etanercept plus methotrexate and DMARD [hydroxychloroquine or sulfasalazine] plus methotrexate), which may have been modified at the start of the extension. All patients who took at least one dose of study drug in the extension phase are included in the extension analyses.
The number of treatment-emergent adverse events per patient-year (PY) was assessed in patients exposed to etanercept in the study, from the start of the initial randomized phase for those in the etanercept-plus-methotrexate group and from the start of the extension for those in the conventional DMARD-plus-methotrexate group. The last observation carried forward approach was used for analyses of clinical efficacy, PROs, and work productivity. Observed cases were included in analyses of the physician and patient satisfaction and CBRU questionnaires.
RESULTS
Patient Disposition and Baseline Characteristics
Of 424 randomized patients evaluated in the first phase of the study, 398 (94%) completed that phase and 386 (91%) entered the optional extension. Of the latter 386 patients, 260 patients had been randomized to receive etanercept plus methotrexate and 126 patients to receive the conventional DMARD plus methotrexate in the first phase of the study. Twenty-four patients (6%) were discontinued during the first 52 weeks of the extension (see Fig. 1, Supplementary Content 1). Of the 361 patients (94%) who continued after week 76 of the extension (1 additional patient completed the first 52-week extension period but discontinued before entering the second 52-week extension period), 331 patients (92%) completed the study: 218 patients (90%) from the randomized etanercept-plus-methotrexate group, and 113 (94%) from the conventional DMARD-plus-methotrexate group in the first phase of the study.
Demographic and baseline characteristics of patients who entered the extension.
| Etanercept + Methotrexate (n=260) |
DMARD + Methotrexate (n=126) |
|
|---|---|---|
| Demographic and Baseline Characteristics | ||
| Age, mean (SD), y | 48.4 (11.8) | 48.4 (11.2) |
| Female, n (%) | 227 (87.3) | 115 (91.3) |
| Ethnicitya | ||
| White, n (%) | 120 (46.2) | 55 (43.7) |
| Mestizos, n (%) | 57 (21.9) | 32 (25.4) |
| African-Latin American, n (%) | 36 (13.8) | 22 (17.5) |
| Other, n (%) | 47 (18.1) | 17 (13.5) |
| Body mass index, (kg/m2) mean (SD) | 26.4 (4.9) | 27.3 (5.0) |
| Prior tobacco use, n (%) | 63 (24.2) | 30 (23.8) |
| Baseline Disease Characteristics | ||
| Disease duration, mean (SD), y | 7.8 (6.9) | 9.0 (7.7) |
| C-reactive protein (mg/L), mean (SD) | 21.4 (26.1) | 20.4 (21.9) |
| Rheumatoid factor positive, n (%) | 227 (87.3) | 104 (82.5) |
| Cyclic citrulinated peptide antibody positive, n (%) | 232 (89.2) | 107 (84.9) |
| Erythrocyte sedimentation rate (mm/h), mean (SD) | 43.1 (16.6) | 42.7 (16.1) |
| DAS28 (ESR), mean (SD) | 6.6 (0.8) | 6.7 (0.8) |
| Tender joint count, mean (SD)b | 25.3 (11.8) | 27.1 (12.6) |
| Swollen joint count, mean (SD)b | 18.4 (8.5) | 19.8 (10.5) |
| Physician global assessment (1–10), mean (SD) | 6.7 (1.6) | 6.7 (1.6) |
| Duration of morning stiffness, min, mean (SD) | 182.5 (265.4) | 142.7 (149.6) |
| Prior Medication Use | ||
| Prior methotrexate use, n (%) | 260 (100.0) | 126 (100.0) |
| Prior NSAID use, n (%) | 216 (83.1) | 110 (87.3) |
| Prior corticosteroid use, n (%) | 194 (74.6) | 93 (73.8) |
| Baseline Patient-Reported Characteristics | ||
| Total HAQ disability index, mean (SD) | 1.6 (0.7) | 1.6 (0.7) |
| Subject global assessment (1–10), mean (SD) | 7.1 (2.0) | 7.2 (1.9) |
| VAS general health (0–100 mm), mean (SD) | 59.7 (21.4) | 61.6 (20.5) |
| VAS pain (0–100 mm), mean (SD) | 64.9 (21.2) | 64.9 (21.5) |
| VAS fatigue (0–100 mm), mean (SD) | 55.7 (26.0) | 58.1 (25.6) |
| SF-36 PCS, mean (SD) | 30.4 (7.2) | 30.1 (6.8) |
| SF-36 MCS, mean (SD) | 40.2 (11.1) | 39.7 (10.1) |
| SF-36 vitality domain score, mean (SD) | 12.3 (3.8) | 12.2 (3.8) |
DAS-28 indicates disease activity Score in 28 joints; DMARD, disease-modifying anti-rheumatic drug; ESR, erythrocyte sedimentation rate; HAQ, health assessment questionnaire; MCS, mental component summary; NSAID, nonsteroidal anti-inflammatory drug; PCS, physical component summary; SD, standard deviation; SF-36-item, short form health survey; VAS, visual analog scale.
a Case report forms included Asian and Mulatto ethnicities as options, but no patients selected these options.
b Values for tender and swollen joint counts are prorated.
Demographic and baseline disease characteristics of patients who entered the 104-week extension are shown in Table 1. Overall, the mean (standard deviation [SD]) age of patients was 48.4 (11.6) and the majority (89%) were female; 45% of patients were white, 23% Mestizo, and 15% African-Latin American. The mean (SD) disease duration was 8.2 (7.2) years. Before entering the study, all patients had been treated with methotrexate, and most had been treated with nonsteroidal anti-inflammatory drugs (85%) and corticosteroids (74%).
Changes in Treatment Regimen
As previously described, investigators were permitted to modify patients’ treatment regimens at the start of the 104-week extension. Few changes were made to the treatment regimens of patients who received etanercept plus methotrexate in the first study phase: 259/260 patients in this group continued to receive etanercept in the extension (see Table 1, Supplementary Content 2). The additional patient in this treatment group had been assigned to etanercept-plus-methotrexate treatment for the randomized phase but was given sulfasalazine in error; this patient’s treatment was subsequently changed from sulfasalazine to etanercept at the beginning of the extension. All patients in this group continued to receive methotrexate. Hydroxychloroquine was added to the treatment regimen in eight patients (3%) and sulfasalazine was added in three patients (1%) in the extension.
Summary of treatment-emergent adverse events in patients exposed to etanercept in the study.
| Adverse Event | No. of Events (Events per PY) |
|---|---|
| Totala (N=365) |
|
| Cumulative Etanercept Exposure=798.11 PY | |
| Total adverse events | 1,354 (1.70) |
| Infections and infestations | 471 (0.59) |
| Neoplasmsb | 17 (0.02) |
| Most frequent adverse eventsc Bronchitis Influenza Nasopharyngitis Urinary tract infection Pharyngitis Nausea Headache Hypertension Sinusitis Back pain Cough |
58 (0.07) 50 (0.06) 56 (0.07) 45 (0.06) 38 (0.05) 29 (0.04) 27 (0.03) 28 (0.04) 19 (0.02) 21 (0.03) 19 (0.02) |
| Total serious adverse events | 57 (0.07) |
| Serious infections and infestations Appendicitis Bronchopneumonia Dengue fever Latent tuberculosis Perirectal abscess Pneumonia Postoperative abscess Postoperative wound infection Septic shock Urinary tract infection |
15 (0.02) 2 (0.003) 1 (0.001) 2 (0.003) 1 (0.001) 1 (0.001) 2 (0.003) 1 (0.001) 1 (0.001) 1 (0.001) 3 (0.004) |
| Serious neoplasmsb Basal cell carcinoma Breast cancer Cervix carcinoma Malignant lung neoplasm Ovarian tumor Prostate cancer |
6 (0.01) 1 (0.001) 1 (0.001) 1 (0.001) 1 (0.001) 1 (0.001) 1 (0.001) |
DMARD indicates disease-modifying anti-rheumatic drug; PY, patient-year.
a Events were calculated for all patients exposed to etanercept from the start of the randomized phase in the etanercept-plus-methotrexate group and from the start of the extension in the DMARD-plus-methotrexate group.
b Benign, malignant, and unspecified.
c Excluding injection site reactions.
In contrast, the treatment regimen was changed at the beginning of the extension in most patients who had been randomized to receive conventional DMARD plus methotrexate in the first phase of the study, with etanercept added as a treatment in 105/126 patients (83%). Hydroxychloroquine was discontinued in 35 patients (55%) and sulfasalazine was discontinued in 51 patients (82%). Methotrexate was continued in all 126 patients in the group.
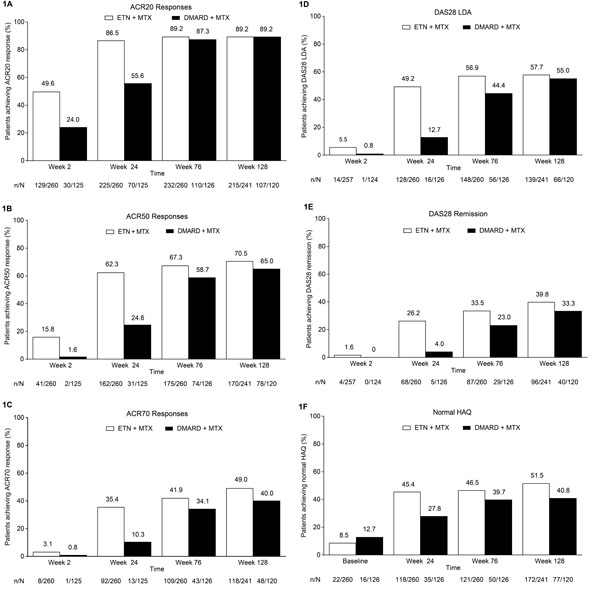
Proportion of patients achieving (A) ACR20 response, (B) ACR50 response, (C) ACR70 response, (D) DAS28 LDA, (E) DAS28 remission, and (F) normal HAQ (≤0.5). Treatment modification was permitted at the start of the extension (i.e., after week 24); during the extension, 259/260 patients in the ETN + MTX group continued to receive etanercept and 105/126 patients in the DMARD + MTX group received etanercept. ACR, American College of Rheumatology; DAS28, Disease Activity Score in 28 joints; DMARD, disease-modifying antirheumatic drug; ETN, etanercept; HAQ, Health Assessment Questionnaire; LDA, low disease activity; LOCF, last observation carried forward; MTX, methotrexate. Analyses included patients who received at least one dose of study drug in the extension phase; LOCF.
Safety
Because the majority of patients in the extension phase were treated with etanercept, safety findings are reported for all patients exposed to etanercept in the study. The total exposure to etanercept during the 24-week randomized phase and the 104-week extension (when treatment assigned in the randomized phase could be modified to include any combination of etanercept, methotrexate, hydroxychloroquine, and/or sulfasalazine) was 798.11 PYs. Among 365 patients exposed to etanercept, 1354 adverse events were reported, which represents 1.70 events per PY (Table 2). The most frequently reported adverse events (excluding injection site reactions) were bronchitis (0.07 events per PY), nasopharyngitis (0.07 events per PY), influenza (0.06 events per PY), urinary tract infection (0.06 events per PY), and pharyngitis (0.05 events per PY).
Fifty-seven serious adverse events were reported in patients exposed to etanercept during the study (0.07 events per PY). Fifteen serious infections (0.02 events per PY) were reported. Six patients had malignancies (0.01 events per PY), which all resulted in withdrawal from the study; two of the six malignancies were considered by the investigators to be treatment-related (i.e. basal cell and cervical cancers). No cases of demyelinating disease or tuberculosis were reported during the study. Adverse events resulted in death in three patients during the study extension, including one death caused by septic shock, respiratory acidosis, and respiratory failure (considered to be treatment-related), one caused by myocardial infarction, and one caused by malignant lung neoplasm (the latter two deaths were not considered treatment-related).
Safety findings in the etanercept-plus-methotrexate and conventional DMARD-plus-methotrexate treatment groups in the second study phase (see Table 2, Supplementary Content 3) were similar to those reported in previous studies of etanercept in RA, with no new safety signals observed.
Clinical Efficacy and PROs
In patients who had been initially randomized to the etanercept-plus-methotrexate group, rates of ACR response and DAS28 LDA and remission were maintained from week 24 to week 128; in those initially randomized to the conventional DMARD-plus-methotrexate group, these rates increased after treatment modification was allowed at week 24 to week 76 and subsequently remained stable through week 128 (Fig. 1A-E). Patients in the randomized etanercept-plus-methotrexate group achieved improvement in DAS28 from baseline to week 24 (mean change [SD], -3.2 [1.3]), which was maintained to week 76 (-3.4 [1.3]) and week 128 (-3.4 [1.4]) (Fig. 2A). Patients in the randomized conventional DMARD-plus-methotrexate group had corresponding DAS28 changes of -1.8 (1.4), -3.4 (1.3), and -3.5 (1.3). Similarly, from week 24 to week 76, relatively small changes were seen in other clinical outcomes in the etanercept-plus-methotrexate group and greater improvement in the DMARD-plus-methotrexate group, followed by stabilization to week 128 (Table 3).
Clinical and patient-reported outcomes in the extension (LOCF).
| Week 76* | Week 128* | |||
|---|---|---|---|---|
| Etanercept + Methotrexate | DMARD + Methotrexate | Etanercept + Methotrexate | DMARD + Methotrexate | |
| n=260 | n=126 | n=241 | n=120 | |
| Clinical Endpoints/Outcomes | ||||
| Tender joint count, mean change (SD) | -21.1 (12.4) | -22.9 (12.8) | -21.4 (12.1) | -24.0 (13.3) |
| Swollen joint count, mean change (SD) | -16.6 (9.0) | -17.7 (10.3) | -16.6 (9.0) | -18.5 (11.2) |
| Physician global assessment, mean change (SD) | -5.2 (2.0) | -5.0 (1.8) | -5.2 (2.1) | -5.2 (2.0) |
| Duration of morning stiffness, mean change (SD) | -98.8 (202.6) | -91.5 (181.7) | -118.8 (294.7) | -90.9 (206.6) |
| Patient-Reported Outcomes | ||||
| Subject global assessment, mean change (SD) | -3.9 (3.0) | -3.4 (2.8) | -4.3 (2.7) | -3.8 (2.8) |
| VAS general health (0-100 mm), mean change (SD) | -33.3 (28.4) | -33.1 (27.1) | -36.3 (27.1) | -36.4 (27.6) |
| VAS pain (0-100 mm), mean change (SD) | -40.4 (28.9) | -37.2 (27.1) | -42.0 (28.6) | -40.6 (26.8) |
| VAS fatigue (0-100 mm), mean change (SD) | -29.4 (31.5) | -28.2 (28.2) | -30.5 (31.3) | -30.4 (30.7) |
* Treatment modification was permitted at the start of the extension (i.e., after week 24); during the extension, 259/260 patients in the ETN + MTX group continued to receive etanercept and 105/126 patients in the DMARD + MTX group received etanercept.
DMARD, disease-modifying anti-rheumatic drug; LOCF, last observation carried forward; SD, standard deviation; VAS, visual analog scale.
Comparable trends were observed in PROs in these treatment groups in the extension, including the proportions of patients achieving a normal HAQ-DI score (Fig. 1F). Minimal changes were seen in the proportions of patients in the etanercept-plus-methotrexate group who achieved MCID improvement in the SF-36 MCS (59%-58%), PCS (76%-77%), and vitality domain (44%-42%) from week 24 to week 76, whereas larger changes were observed in the proportions of patients in the conventional DMARD-plus-methotrexate group achieving these endpoints (46%-59%; 59%-73%; and 27%-38%, respectively). Rates of MCID improvement in these scores subsequently remained at stable levels to week 128 in both groups. As for clinical outcomes, minimal changes from week 24 to week 76 were reported for PROs, including HAQ-DI, the SF-36 MCS and PCS, and vitality domain scores, among patients initially randomized to receive etanercept plus methotrexate, with greater improvement after week 24 seen among those randomized to receive hydroxychloroquine or sulfasalazine plus methotrexate (Fig. 2B-E; Table 3). Improvements were maintained from week 76 to week 128 in PROs in both treatment groups.
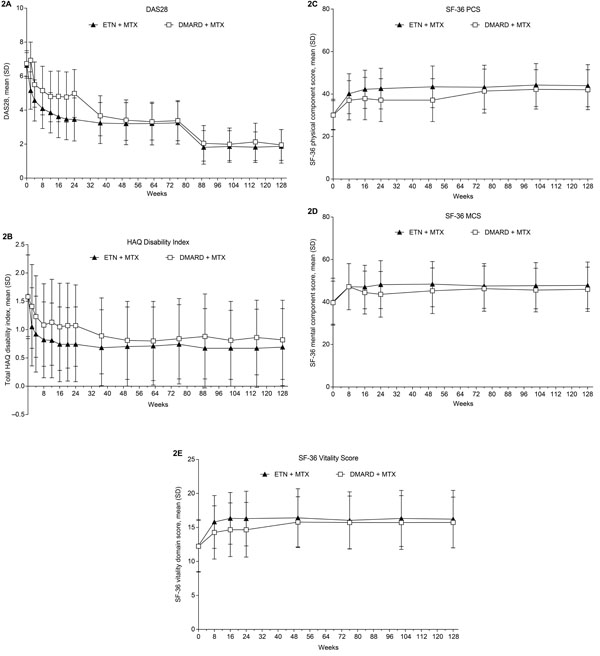
Mean scores (SD) for (A) DAS28, (B) total HAQ Disability Index, (C) SF-36 PCS, (D) SF-36 MCS, and (E) SF-36 vitality domain. Treatment modification was permitted at the start of the extension (i.e., after week 24); during the extension, 259/260 patients in the ETN + MTX group continued to receive etanercept and 105/126 patients in the DMARD + MTX group received etanercept. DAS28, Disease Activity Score in 28 joints; DMARD, disease-modifying anti-rheumatic drug; ETN, etanercept; HAQ, health assessment questionnaire; MCS, mental component summary; MTX, methotrexate; PCS, physical component summary; SF-36, 36-item short-form health survey. Analyses included patients who received at least 1 dose of study drug in the extension phase; LOCF.
Satisfaction Questionnaire
The proportion of physicians who were satisfied with control of their patients’ RA remained stable (at 94%-96%) for patients in the etanercept-plus-methotrexate group from week 24 to week 128; the physician satisfaction rate increased from 42% to 98% for patients in the conventional DMARD-plus-methotrexate group between these time points. No change was seen in the proportion of patients in the etanercept-plus-methotrexate group who were willing or very willing to retake the RA treatment received in the past 6 months from week 24 to week 76 (99%), whereas an increase was reported in the proportion of willing or very willing patients in the DMARD-plus-methotrexate group in this timeframe (82%-98%). Minimal change was seen in patient satisfaction after week 76 in either group.
Work Productivity and Resource Utilization
Based on responses to the WPAI:RA questionnaire, patients in both treatment groups had improvement from baseline to week 24 in the percentage of work time missed and the overall work impairment due to RA in the previous 7 days, and this improvement was maintained through the 104-week extension (Fig. 3A and B). Minimal changes were seen in current employment, according to responses to the CBRU questionnaire (Fig. 3C). In both treatment groups, reductions were seen from baseline to week 24 in the proportion of patients who required visits to a rheumatologist or the emergency department in the past 6 months, and in those who required caregiver assistance in the past month (Fig. 3D-F); these rates generally remained stable through the extension.
DISCUSSION
At the start of the extension of this study of RA patients from the Latin American region, modifications in randomized phase treatment regimens were permitted according to study protocol. Few changes were made to the treatment regimens of patients originally randomized to receive etanercept-plus-methotrexate therapy. Only 11 patients (4%) in this group had their initial treatment regimen supplemented with a conventional DMARD. In contrast, the treatment regimen of most patients originally randomized to receive a conventional DMARD plus methotrexate (105 patients; 83%) was modified by replacing the conventional DMARD with etanercept or adding etanercept to DMARD therapy. Overall, 365 patients (95%) were receiving etanercept plus methotrexate in the extension.
The safety profile of etanercept in the 104-week extension phase of this study remained consistent with that observed in previous studies of etanercept in RA, with no new safety signals reported. No marked changes in the incidence, type, or severity of adverse events were identified with continued exposure to etanercept in patients randomized to receive combination etanercept-plus-methotrexate therapy in the initial phase or with the addition of etanercept in those randomized to combination DMARD-plus-methotrexate therapy. Continuation of the combination biologic-plus-methotrexate treatment through 104 weeks in patients initially assigned to that treatment group resulted in consistently good clinical responses and PROs. The addition of etanercept in the extension to the randomized DMARD-plus-methotrexate treatment regimen resulted in outcomes that approached or were similar to those seen in the randomized etanercept-plus-methotrexate group in the first phase for most assessments. Improvements from baseline were seen after treatment was initiated in work productivity, caregiver burden, and resource utilization, which remained largely stable throughout the 104-week extension in both groups.
In Latin America, as elsewhere, DMARDs are recommended as first-line therapy in patients with RA depending on factors such as disease activity and response to prior treatment. Biologic agents are generally not initiated early in the disease course because of their high costs and current labeling that preclude their use until patients have failed to respond to methotrexate or other DMARDs. Multiple DMARDs in combination are frequently prescribed for patients with high disease activity or who have failed to respond to maximum doses of methotrexate monotherapy; however, many patients treated with combination DMARD therapy do not achieve the desired treatment targets of clinical remission or at least LDA. In this study, note that the majority of patients who had been treated with a conventional DMARD plus methotrexate for 24 weeks were switched to combination therapy including etanercept for the 104-week extension. These findings suggest that most physicians attempted to further improve responses of patients initially treated with a conventional DMARD-plus-methotrexate combination in the first study phase with the addition of etanercept. The outcomes reported at weeks 76 and 128 of this study appeared to confirm the benefits of this strategy.
Results of this study, which suggest that combination etanercept-plus-methotrexate therapy is clinically superior to conventional DMARD-plus-methotrexate therapy [12], contrast with those of previous reports from the TEAR and RACAT studies in which biologic-plus-methotrexate and triple DMARD regimens resulted in comparable clinical outcomes [13, 14]. The findings of this regional study are more in keeping with those of the SWEFOT study, which showed a significantly greater clinical benefit with infliximab-plus-methotrexate therapy versus triple DMARD therapy [15]. However, it cannot be overlooked that marked differences in the designs and population characteristics of these studies preclude a meaningful comparison of findings. In a more judicious comparison, the safety and efficacy profile of etanercept observed in the 104-week extension phase of the current study is consistent with that found in other open-label long-term extension studies of etanercept-treated patients with RA [16-19].
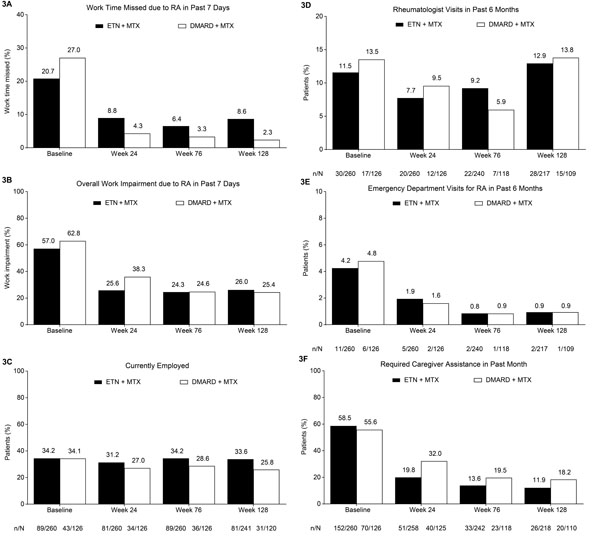
The percentage of (A) work time missed and (B) overall work impairment due to RA in the past 7 days based on patient responses on the WPAI:RA questionnaire. Proportions of patients who are (C) currently employed; who have required (D) a rheumatologist visit or (E) an emergency department visit in the past 6 months; and who have required (F) caregiver assistance in the past month based on responses on the CBRU questionnaire. Treatment modification was permitted at the start of the extension (i.e., after week 24); during the extension, 259/260 patients in the ETN + MTX group continued to receive etanercept and 105/126 patients in the DMARD + MTX group received etanercept. Analyses included patients who received at least one dose of study drug in the extension phase. WPAI:RA and CBRU employment findings analyzed using LOCF; all other CBRU findings based on observed cases. CBRU, caregiver burden and resource utilization questionnaire; DMARD, disease-modifying anti-rheumatic drug; ETN, etanercept; LOCF, last observation carried forward; MTX, methotrexate; RA, rheumatoid arthritis; WPAI:RA, work productivity and activity impairment: rheumatoid arthritis questionnaire.
The open-label design of the study may be considered a limitation. Patients were required to satisfy fixed eligibility criteria; as a result, findings from this study population may not be applicable to those observed in the general RA population. In addition, some patients were able to receive treatment not covered by a payer and thus financially unavailable in the clinical practice setting. The number of patients evaluated was too small to allow subanalyses of treatment efficacy of the nonbiologic DMARDs at varying dosages. Finally, because the population studied did not include patients from all Latin American countries and ethnic groups, the findings and conclusions may not be generalizable to other patients in the region.
Although regional guidelines in Latin America generally recommend that biologics be withheld until patients have shown an inadequate response to at least two conventional DMARD regimens, increasing evidence suggests that early aggressive treatment may be warranted to improve achievement of remission or LDA and limit joint damage and functional deficits. The findings of this study support the early use of etanercept plus methotrexate in patients from Latin America who had active RA despite previous methotrexate therapy. Additional studies in this region may be warranted to further explore the consequences of early versus delayed introduction of biologic therapy in RA.
CONCLUSION
This report provides findings from a 2-year extension of a randomized open-label trial conducted exclusively in Latin America to evaluate safety and efficacy outcomes with physician-selected treatment (i.e. a combination of etanercept plus methotrexate or conventional nonbiologic DMARD plus methotrexate) in patients with moderately to severely active RA despite MTX therapy. The findings indicate that when a change in treatment regimen was permitted, most physicians did not alter the biologic-plus-methotrexate regimen but added etanercept to the conventional DMARD-plus-methotrexate regimen (in addition to or instead of the conventional DMARD). No new safety signals were reported during the study; the adverse event profile was consistent with that observed in previous clinical trials of etanercept. The favorable clinical responses and PROs achieved in the initial randomized phase were maintained in the extension phase among patients who continued to receive the biologic-plus-methotrexate regimen. Most outcomes among patients in the randomized DMARD-plus-methotrexate group improved in the extension phase after addition of etanercept to the treatment regimen.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher's Web site along with the published article.
CONFLICT OF INTEREST
Daniel A. Machado, Ricardo M. Xavier and J. Abraham Simon have received compensation for advisory boards and speaker fees from Pfizer. RG has received consulting fees from Pfizer. Linda Mele, Qi Shen, Ronald Pedersen, Sameer Kotak and Bonnie Vlahos were employees of Pfizer during this study and manuscript preparation.
Source of Funding
The Latin American RA study was funded by Wyeth, which was acquired by Pfizer in October 2009. Medical writing support was provided by Donna McGuire, CMPP, of Engage Scientific Solutions and was funded by Pfizer.
ACKNOWLEDGEMENTS
The authors thank the investigators of this study (listed below).
|
Argentina Juan Carlos Barreira Carlos Baruzzo Alberto Berman Maria Celina de la Vega Ruth Celina Moriondo de Pizzolato Javier Duhau Julio Hofman Eleonora Lucero Daniel Augusto Machado Osvaldo Daniel Messina Jose Luis Christian Moreno Eduardo Fabian Mysler Horacio Venarotti |
Chile Pedro Miranda Oscar Neira Colombia Monique R. Chalem Mario Enrique Diaz William Jose Otero Escalante Elias Gonzalo Forero Edwin Antonio Jauregui John Dario Londono Jose Fernando Molina Juan Jose Jaller Raad Diego Luis Saaibi Solano Edgardo David Tobias |
Mexico Federico Galvan Jesus Abraham Simon Cesar Pacheco-Tena Panama Generoso Guerra Antonio Cachafeiro Vilar |

