All published articles of this journal are available on ScienceDirect.

Anti-MDA5 Antibody Dermatomyositis Overlap with Systemic Lupus Erythematosus: A Case Report and Review of the Literature
Abstract
Background:
Dermatomyositis (DM) is an autoimmune connective tissue disease that primarily targets the muscle, skin, and lungs. Many patients have autoantibodies that correspond to distinct clinical phenotypes. Melanoma differentiation-associated gene 5 (anti-MDA5) antibody, a specific antibody that targets the melanoma differentiation-associated gene 5 (MDA5), has been reported in DM cases and is significant for a distinct cutaneous presentation and rapidly progressive interstitial lung disease.
Objective:
Herein, we describe a patient with DM with a positive anti-MDA5 antibody and characteristic clinical phenotype, who subsequently developed coexisting systemic lupus erythematosus (SLE). A diagnosis of SLE was supported by his clinical phenotype, positive serologies, hypocomplementemia, and progression to glomerulonephritis and lupus cerebritis, features of which fulfilled the American College of Rheumatology criteria for SLE.
Conclusion:
DM is known to overlap with other autoimmune diseases, including SLE, and coexistence can lead to a wide variety of clinical presentations. SLE overlapping with anti-MDA5 positive DM may present with distinct clinical features.
INTRODUCTION
Dermatomyositis (DM) is an autoimmune connective tissue disease that primarily targets the muscle, skin, and lungs. Many patients have autoantibodies that correspond to distinct clinical phenotypes. Herein, we describe a patient with DM and a positive melanoma differentiation-associated gene 5 (anti-MDA5) antibody, who subsequently developed coexisting systemic lupus erythematosus (SLE).
REPORT OF A CASE
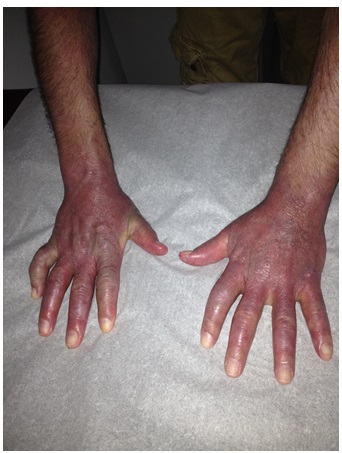
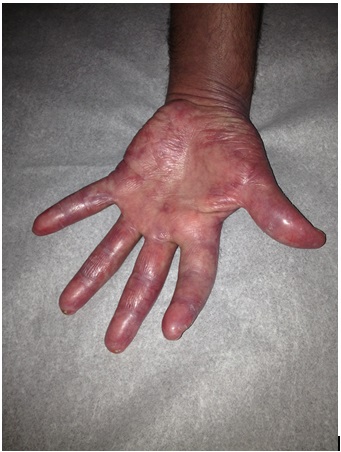
A Caucasian man in his 40s presented with 4 months of periorbital erythema and swelling, symmetric arthralgia of the hands and feet, and proximal muscle weakness bilaterally. Physical exam was significant for edema of the face and hands, violaceous macules and papules on the palms, erythema over the metacarpophalangeal (MCP) joints, and calcinosis cutis (Figs. 1 and 2). He had inflammatory arthritis involving the wrists, MCP, and proximointerphalangeal (PIP) joints. Laboratory values were significant for a normal creatine phosphokinase (CPK) level (84 U/L; normal: 44-196 U/L) and a highly elevated aldolase (19.5 U/L; normal: 1.5-8.1 U/L). Other serologies, including direct antinuclear antibody (ANA), rheumatoid factor, anti-dsDNA, anti-Sm, anti-Jo1, anti-SCL70, myeloperoxidase and proteinase 3 were negative. The patient’s creatinine was 0.94 mg/dL and urinalysis was normal. A muscle biopsy was not obtained.
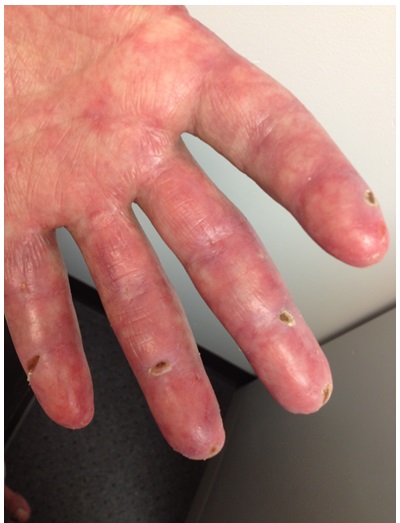
A diagnosis of hypomyopathic dermatomyositis was suspected. A second ANA, this time by immunofluorescence assay (IFA), was found to be positive (1:80, speckled pattern). A workup for underlying malignancy was unremarkable. The patient began treatment with prednisone and methotrexate, with ensuing resolution of his weakness, improvement of arthritis, and normalization of his aldolase. However, skin findings persisted. Ultimately, his palmar and digital lesions progressed to ischemic lesions in the fingertips (Fig. 3) and small ulcers in the dorsum of his MCP and PIP joints and the soles of both feet. Courses of azathioprine and, later on, courses of mycophenolate mofetil and dapsone were added to his preexisting regimen of methotrexate, resulting in improvement in arthritis. However, he then developed hyperkeratotic pustules on his palms, digital discoloration consistent with chilblains, and progression of the digital ulcers. His course was complicated by septic arthritis of the PIPs, requiring several courses of antibiotics.
The patient demonstrated a moderate positive test for anti-MDA5 (CADM-140) detected by immunoprecipitation, supporting the diagnosis of anti-MDA5 DM. He denied shortness of breath. Pulmonary function tests (PFTs) reported minimal obstructive lung defects, a moderate decrease in diffusing capacity, and normal lung volumes. A subsequent high resolution CT revealed thickening of the interlobular septa, suggestive of an interstitial process.
Approximately 1 year after initial presentation, routine laboratory workup revealed a positive ANA by IFA (titer of 1:1280, centromere pattern), positive anti-dsDNA (48 IU/mL; normal < 4 IU/mL), low C3 (46 mg/dL; normal 90-180 mg/dL), and normal C4 (17 mg/dL; normal 16-47 mg/dL). Anti-smooth muscle and anti-RNP remained normal. The patient developed new 3+ hematuria and 2+ proteinuria on urinalysis, with creatinine elevated to 1.26-2.1 mg/dL. Soon after, a kidney biopsy revealed World Health Organization (WHO) class II glomerulonephritis with tubulointerstitial inflammation and positive immunofluorescence in the mesangium (2+ for lgG, 1+ for IgM, trace for IgA, 2+ for C3, 2+ for C1q and 2+ for kappa and lambda) and the tubular basement membrane (2+ for IgG, 1+for lgM, 1+for C3, 2+ for Clq and 2+ for kappa and lambda). Given the clinical, serologic, and renal findings, a diagnosis of probable DM and SLE overlap was made. Because of the recent infections and the concern for MTX-induced interstitial lung disease in a potentially susceptible host, the patient’s treatment was changed to intravenous immunoglobulin (IVIG). He received 5 doses of IVIG, totaling 375 grams over 4 months. His cutaneous involvement and urinary sediment improved, and his complement normalized.
Six months later, the patient developed altered mental status, paucity of speech, and left facial droop. Brain MRI revealed abnormal enhancements consistent with acute infarcts and breakdown of the blood-brain barrier, suggestive of an underlying vasculopathy. There was no clinical or laboratory evidence of infection. An extensive laboratory evaluation was significant for hypocomplementemia, weakly positive anti-dsDNA antibody, and a positive anti-centromere IgG antibody (59 Au/mL; normal 0-40 Au/mL). A subsequent brain biopsy of the right frontal lobe showed microscopic sub-acute infarcts in the white matter and disrupted cortical vessels, consistent with prior vasculitis and, given the concurrent clinical and laboratory findings, specifically concerning for lupus cerebritis. No active vasculitis was noted, and a subsequent coagulopathy workup was negative, including negative cardiolipin IgG and IgM antibodies and negative lupus anticoagulant. The patient was treated with pulse solumedrol as an inpatient, followed by oral prednisone 1 mg/kg, and 6 monthly IV cyclophosphamide pulses at a dose of 1.8 grams. Mental status improved, complement normalized, and proteinuria eventually resolved.
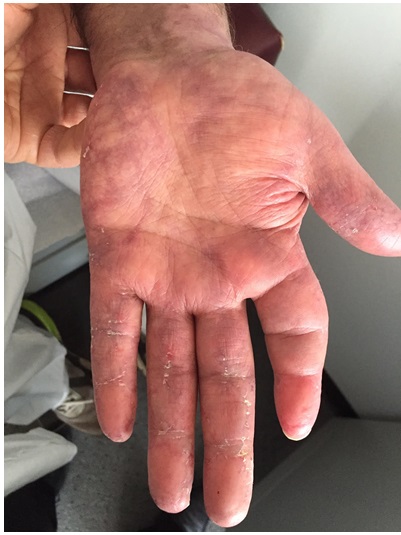
One year after the lupus cerebritis episode he developed severe headache, nausea, lethargy, and difficulty in moving. A CT scan revealed a subdural hematoma within the posterior fossa with compression of the fourth ventricle. The patient had no history of fall or trauma. A CT angiogram revealed preexisting white matter ischemia and multiple areas of intracranial stenosis, with high-grade narrowing or occlusion of portions of the middle cerebral artery and reconstitution via small collateral vessels indicative of a preexisting process. The findings were suggestive of vasculitis or other vasculopathy in the setting of multiple prior infarcts. Urinalysis showed proteinuria and moderate blood, though complement remained normal. Since then, the patient has been treated with mycophenolate mofetil 3 grams daily, hydroxychloroquine 200 mg twice daily, and prednisone 5 mg daily. He exhibits moderate cognitive impairment and poor functional capacity compared to his baseline prior to presentation. He otherwise feels well, with the exception of pain in the joints of his hands, as well as purplish discoloration of the hands, areas of chilblain in the palms, and keratotic, fissured changes of the PIPs and DIPs (Fig. 4). Complement and urinary sediments have remained normal and his ANA by IFA was most recently 1:40 (centromere pattern) with a negative anti-dsDNA.
DISCUSSION
Anti-MDA-5, a specific antibody that targets the melanoma differentiation-associated gene 5 (MDA5), has been reported in DM cases with a distinct clinical presentation and prognosis [1].
MDA-5 is a cytoplasmic RNA helicase that belongs to the retinoic acid-inducible gene-I (RIG-I) family [2]. MDA 5 and RIG-I function as pathogen recognition receptors against viral double stranded RNA and, together, these two molecules may play an important role in innate immune reactions against viral infection [3]. Reported rates of MDA-5 autoantibody detection among DM patients range from 13% to 35% [4-7].
The clinical characteristics of DM with anti-MDA5 consist of significantly low or normal CK, a characteristic cutaneous phenotype, and higher risk of interstitial lung disease. Anti-MDA5 DM patients are typically ANA negative and have an isolated elevation of aldolase but normal CPK, as seen in our patient [5]. Anti-MDA5 positive patients have distinctive erythematous macules and papules on the palms and soles, occasionally associated with hyperkeratosis [5]. Twenty-eight percent of patients have cutaneous ulcers, with 56% occurring over the extensor surfaces of the joints, 42% at the digital pulp or periungual areas, and 58% located elsewhere [8]. Patients with anti-MDA5 can also have tender, swollen gums or oral erosions.
The patient we described had significant violaceous discoloration in the hands resembling chilblain lupus, pustules on the palms and soles, and ulcers over the MCPs and PIPs that were difficult to control and eventually caused septic arthritis. It has been described that patients with anti-MDA5 have a high prevalence of interstitial lung disease (60-100%) [9]. Our patient had changes in his PFT and high resolution CT suggestive of interstitial lung disease, but he was asymptomatic.
The ideal treatment criteria for anti-MDA5 patients has not yet been established. In a study where the patients were divided according to the anti-MDA5 titer, the patients with high antibody levels (>500 U/ml) did not respond to high dosage of glucocorticoids (>2 mg/kg/day) combined with cyclophosphamide, whereas patients with lower levels of anti-MDA5 did respond to glucocorticoids combined with cyclophosphamide or cyclosporine, both in the lung function and skin ulcers [10]. MTX was not explicitly mentioned as a treatment option in this study, likely owing to its association with interstitial lung disease. Our patient did not respond to steroids or multiple cytotoxic medications, however he improved with IVIG.
In our patient, anti-MDA5 positive DM preceded a new, overlapping diagnosis of SLE, per the American College of Rheumatology (ACR) diagnostic criteria. One year after the diagnosis of DM, he was found to have a positive ANA (but, centromere pattern) and positive anti-DNA double stranded, low complement, and abnormal urinary sediment (hematuria 2+ and proteinuria 1+). A kidney biopsy revealed WHO class II SLE glomerulonephritis with tubulointerstitial inflammation and positive immunofluorescence in the mesangium and tubular basement membrane. Glomerular involvement is rare in dermatomyositis. It has been suggested that there are 2 possible mechanisms for renal damage in these patients: tubular disorder related to myoglobulinemia and myoglobinuria and, less frequently, chronic glomerulonephritis. There are also cases reported with membranous and mesangial proliferative glomerulonephritis [11].
DM is known to overlap with other autoimmune diseases, including SLE, and coexistence can lead to a wide variety of clinical presentations. While the coincidence of anti-MDA5 positive DM and SLE has not been investigated directly, a handful of studies assessed the prevalence of anti-MDA5 antibodies among connective tissue disease patients, failing to identify any cases of anti-MDA5 in SLE cohorts. One study of Chinese patients noted that MDA5 reactivity was detected at a significantly higher frequency among clinically amyopathic DM (CADM) patients than in classic DM patients, but was not detected in patients with polymyositis, SLE, Sjögren’s syndrome, or mixed connective tissue disease, nor was it detected in a limited number of patients with idiopathic pulmonary fibrosis or in healthy controls [10]. A similar study of a Mediterranean cohort showed comparable results, with no evidence of anti-MDA5 positivity in a control group of 25 SLE patients [12, 13].
In our case, the unusual association between dermatomyositis with MDA5 and SLE raises the possibility that the phenotype of dermatomyositis is not only different based on the skin manifestations, poor response to cytotoxics, and potential lung involvement, but could also explain the renal involvement and perhaps the characteristics of central nervous system involvement.
It has been shown that MDA5 can be induced in normal human mesangial cells, and it was possible to detect intense immunoreactivity for MDA5 in renal biopsy specimens obtained from patients with severe lupus nephritis or proteinuric IgA nephropathy. In non-immune complex-mediated diseases, MDA5 was undetectable. De novo synthesis of interferon-beta may mediate the expression of induced MDA5. MDA5 is known to mediate CXCL10 induction in bronchial epithelial cells infected with rhinovirus and CXCL10 has been implicated in the pathogenesis of glomerular nephritis including lupus nephritis [14]. Even though the pathological characteristics in our patient are explained best by lupus nephritis, the fact that he had mainly mesangial proliferation and involvement of the tubular basement membrane, which responded well to IVIG, suggests that the renal involvement may also be related to his DM.
Our patient developed the 2 episodes of CNS involvement consistent with vasculitis or vasculopathy, he did not have active renal disease and the skin involvement in his hands and feet was stable. The possibility that anti-MDA5 may play a role in the etiology of CNS vasculopathy is considered and may also be a unique factor of this overlap.
CONCLUSION
In summary, we report a case of anti-MDA5 DM overlapping with SLE. There is evidence in the literature that anti-MDA5 may have had an impact on the clinical characteristics of DM and SLE. We have discussed some, but further observations are needed to better characterize this association.
AUTHORS’ CONTRIBUTIONS
Drs. Futran and Franks and had full access to all of the case report information in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: N/A
Acquisition, analysis, and interpretation of data: Futran, Franks, Milam.
Drafting of the manuscript: Futran, Franks, Milam.
Critical revision of the manuscript for important intellectual content: Futran, Franks, Milam.
Statistical analysis: N/A
Obtained funding: N/A
Administrative, technical, or material support: N/A
Study supervision: N/A
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

