All published articles of this journal are available on ScienceDirect.

Two Subsets of Large Vessel Vasculitis Characterized by the Absence or Presence of Spondyloarthritis or its Associated Diseases
Abstract
Background:
Whilst large vessel vasculitis (LVV) predominantly occurs in isolation, associations with other infectious and non-infectious diseases have been reported. Limited data describing associations with various autoimmune diseases (AI), including spondyloarthritis exists. The aim of this study was to characterize the association of LVV and spondyloarthritis or its associated diseases (SpAD).
Methods:
A single centre, retrospective study of patients ≥50yrs with first presentation LVV between 01.06.2008-01.06.2015 was performed. Patients were categorized according to SpA or associated disease, other AI or idiopathic LVV (iLVV). Clinical, laboratory and imaging findings were compared. Kaplan-Meyer survival analysis, with relapse taken as the primary end-point, was performed.
Results:
LVV was confirmed in 62 pts, of who 16/62 (26%) had SpA or associated disease. In these patients, LVV presented earlier (59.2 SpAD vs. 68.1 AI and 70.3yrs iLVV; p=0.01) and occurred predominantly in spring compared to autumn and winter in non-SpA patients, was associated with more centralised pattern of distribution (p=0.05) and was more likely to exhibit a refractory course (p=0.05). Overall relapse rates were similar across groups. Smoking-status influenced age of onset in all groups, being associated with earlier onset.
Conclusion:
A clear association between LVV and SpAD exists. LVV associated with SpAD has a particular phenotype characterised by earlier onset, thorax-limited disease and increased risk of a refractory course. Given on-going LVV treatment trials further genetic and pathophysiological characterization appears warranted, to evaluate potential variation in treatment response and optimize future care.
INTRODUCTION
Large vessel vasculitis (LVV) refers to inflammation of the aorta and its main branches. When occurring in isolation, the distinct phenotypes of Takayasu’s arteritis (TA) and giant-cell arteritis (GCA) are assigned based on patient age and patterns of vessel involvement [1]. LVV is also an integral component of well-described syndromes, such as Kawasaki’s and Behçet’s syndromes. Several reports have identified possible associations between various autoimmune diseases, including spondyloarthropathy-associated diseases (SpAD) such as inflammatory bowel disease [2, 3] and ankylosing spondylitis [4, 5]. Registry data has previously highlighted a raised incidence of GCA in patients with psoriasis and psoriatic arthritis patients [6] and in a recent small study, presented clinical evidence supporting a higher prevalence of LVV in patients with psoriatic arthritis and sacroiliitis [7].
The aim of this study was to further assess the relationship between LVV and SpAD. Beyond merely establishing a link between these conditions, the main focus is to characterize LVV in SpAD and compare the findings to both patients with non-SpAD autoimmune disease and those with no known autoimmune disease.
MATERIALS AND METHODS
A retrospective analysis of 1st presentation of large vessel vasculitis at our institution between 1st June 2008 and 1st June 2015 was performed. To avoid overlap with Takayasu arteritis, only patients ≥50 years at symptom onset were included. Diagnosis was confirmed in all cases either by imaging or vessel biopsy. Patients subsequently diagnosed with paraneoplastic vasculitis or severe atherosclerosis were excluded from the analysis.
Patient charts were reviewed, with presenting symptoms and diagnostic parameters being tabulated. The cohort was divided into 3 groups, based on coexisting spondyloarthropathy or associated disease (SpAD), another known autoimmune disease (AI), or iLVV. A diagnosis of SpAD required fulfilment of Assessment of Spondyloarthritis International Society (ASAS) criteria for SpA or its associated clinical features [8-10]. diagnosed by the relevant medical specialists. Other autoimmune diseases required either confirmation of the relevant antibodies or fulfilment of other recognized diagnostic criteria in the absence of clinical features outlined in the ASAS criteria (Supplementary Data Sheet).
Current and treatment prior to LVV development were also collated for all patients, with particular reference to immunosuppressants. Corticosteroid treatment equating to ≥5mg prednisolone daily, or any disease modifying medication was considered significant. Treatment of large vessel vasculitis adhered to EULAR recommendations [11] and comprised of 1mg/kg oral prednisolone (max. 60mg daily) initially in combination with methotrexate (MTX). Patients with contraindications or poor tolerance of MTX received corticosteroids in combination with azathioprine.
Sub-analysis, examining the independent influence of age on disease presentation and course was subsequently performed in 3 groups, taking arbitrary cut-offs of <60yrs for younger patients and >70yrs for older patients.
Data are reported as means (±standard deviation), with time-dependent variables being expressed as median [interquartile ranges]. Unless stated otherwise, p-values are two-sided, with <0.05 being considered statistically significant. Categorical variables were analysed using Chi-Squared test or Fisher´s exact test. Continuous variables were compared using ANOVA, or in the age-dependent sub-analysis with the Kruskal-Wallis Test. Relapse-free survival was calculated using a Kaplan-Meier analysis.
RESULTS
In total 62 patients (48 female; 77%) were included. The diagnosis of LVV was based on imaging in 53 (85.5%) patients and on a pathological result of a biopsy from the temporal artery in 9 (14.5%) patients. Median age at LVV diagnosis was 65.9 [57.6 – 72.1] years. A known autoimmune disease was present in 29 (47%) patients, of whom 16 (26%) had a SpAD. Whilst no differences in gender composition were observed, patients in the latter group were significantly younger at presentation (59.2yrs) versus 68.1yrs among other AI diseases and idiopathic LVV (70.3 yrs; p=0.01). SpAD was evident in over one-third of patients presenting before 70 years, whereas only 1 patient (4%) presenting after 70yrs had SpAD (p=0.05). Detailed information on cohort group composition is illustrated in Fig. (1). Patient demographics are summarized in Table 1.
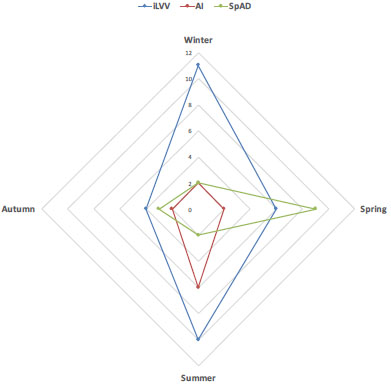
No significant differences in clinical symptomatology of LVV were seen between groups, with myalgia being the commonest symptom in all groups. Among SpAD patients the majority (56%) became symptomatic in spring, whereas autumn and winter predominated in the other groups (Fig. 2), p=0.02). The median interval between symptom onset and diagnosis was 91 [49 – 198] days for the entire cohort, again with no differences between groups.
Raised serum C-reactive protein and erythrocyte sedimentation rates in the absence of leucocytosis were characteristic of all groups with no significant quantitative differences.
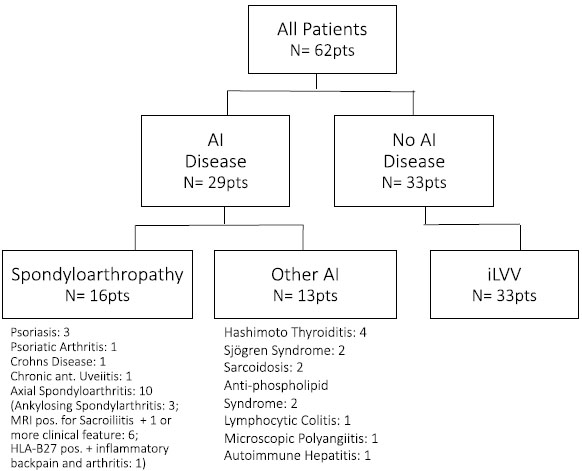
Whilst a slightly lower proportion of the SpAD underwent positron emission tomography (PET-CT) (44% vs. 62% AI and 64% iLVV; p=0.46), no significant imaging bias within the groups was apparent. Whereas the extent of vessel involvement was similar across all groups, significant differences in the patterns of disease existed. SpAD patients demonstrated predominantly central disease with involvement of the ascending aorta in 92% of cases, compared to 83% in the control group and 55% of AI patients (p=0.05). Conversely, involvement of the abdominal aorta among SpAD patients was lower than in the other groups (62% vs. 90% iLVV and 73% AI; p=0.08).
In terms of treatment, no differences in drug management existed between groups (p=0.39). Although no differences in overall relapse rates were observed (p=0.62), SpAD patients were more likely to develop refractory disease, defined as ≥2 relapses on treatment (31% vs. 6% iLVV and 23% AI; p=0.05). Pre-existing treatment with corticosteroids at LVV onset was noted in 8 (13%) patients. Developing LVV whilst taking corticosteroids did not increase the likelihood of relapsing on treatment (p=0.79) or developing refractory disease (p=0.76). None of the patients in the SpAD or AI groups received biological agents or disease-modifying anti-rheumatic drugs (DMARDs). Main treatment was non-steroidal anti-inflammatory drugs (NSAID) or in case of peripheral manifestations low dose prednisolone therapy. All patients developed their underlying condition prior to LVV onset, occurring at a median 79 [12 – 227] months in SpAD patients and 42 [8 – 284] months in patients with AI.
A history of previous or current smoking contributed to LVV development at a younger age (p=0.007). This effect was independent of SpAD status, although impact was greater in non-SpAD patients (SpAD 57.1 vs. 61.1years, non-SpAD 59.3 vs. 70.9years; p=0.004). Similarly, concomitant smoking among SpAD patients had a negative impact on 1-year relapse free survival (83% vs. 56%; p=0.02) but not on overall incidence of relapse (p=0.87) or refractory disease (p=0.27).
DISCUSSION
This paper introduces the coexistence of large vessel vasculitis (LVV) with spondyloarthritis or its associated diseases. Although also described in Behçet’s syndrome and rheumatoid arthritis, LVV most commonly occurs in isolation and is considered as either Takayasu’s or giant-cell arteritis(GCA) [1]. Whilst sharing similar pathogenesis, these are distinguished by age at onset and pattern of disease distribution, with the latter being more common in older populations, with female sex and age >70 years at presentation being typical. Among adults >50yrs, incidence varies between 15-25/100,000 [1]. Exact pathogenesis is unclear, with a multifactorial aetiology combining environmental and genetic factors triggering cellular and humoral immune responses suspected [12]. Tobacco consumption is a good example of the former, given the strikingly higher than anticipated incidence particularly among women 65 years. Its impact on premature onset of LVV onset was-profound. This correlates with recent findings suggesting earlier onset of ANCA associated vasculitides among smokers [13]. Further assessment of the relationship of smoking to large vessel vasculitis in larger cohorts appears warranted.
| iLVV | SpAD | AI | p-value | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient Demographics | |||||||||
| n | 33 | 16 | 13 | ||||||
| Female, n (%) | 26 | (79%) | 11 | (69%) | 11 | (85%) | 0.58 | ||
| Never Smoker, n (%) | 14 | (42%) | 6 | (38%) | 3 | (23%) | 0.41 | ||
| Age at Diagnosis, yrs | 70.3 | [59.3-73.3] | 59.2 | [54.0-63.8] | 68.1 | [58.7-72.3] | 0.01 | ||
| Follow-up, mts | 11 | [3-36] | 38 | [18-63] | 39 | [7-52] | 0.01 | ||
| Clinical Features | |||||||||
| Fever, n (%) | 7 | (21%) | 7 | (44%) | 4 | (31%) | 0.22 | ||
| Weight loss, n (%) | 21 | (64%) | 8 | (50%) | 7 | (54%) | 0.73 | ||
| Night Sweats, n (%) | 16 | (48%) | 9 | (56%) | 6 | (46%) | 0.71 | ||
| Myalgia, n (%) | 22 | (67%) | 9 | (56%) | 7 | (54%) | 0.70 | ||
| Headache, n (%) | 18 | (55%) | 8 | (50%) | 4 | (31%) | 0.34 | ||
| Jaw claudication, n (%) | 10 | (30%) | 2 | (13%) | 3 | (23%) | 0.44 | ||
| Symptom Duration, d | 92 | [53-203] | 97 | [38-201] | 79 | [53-181] | 0.91 | ||
| Laboratory Parameters | |||||||||
| Leucocytes, Tsd/µl | 8.8 | [7.8-10.0] | 9.1 | [8.2-10.7] | 10.6 | [7.5-11.0] | 0.73 | ||
| CRP, mg/l | 90 | [50-120] | 73 | [37-118] | 107 | [31-149] | 0.67 | ||
| ESR, mm/hr | 76 | [69-93] | 79 | [58-88] | 81 | [62-100] | 0.90 | ||
| Imaging | 0.46 | ||||||||
| PET-CT, n (%) | 21 | (64%) | 7 | (44%) | 8 | (62%) | |||
| - SUVmax | 4.4 | [3.2-6.0] | 3.1 | [2.9-3.2] | 3.5 | [3.3-4.7] | 0.305 | ||
| MRI, n (%) | 8 | (24%) | 6 | (38%) | 3 | (23%) | |||
| Site affected | |||||||||
| - Ascending Thoracic Aorta, n (%) | 24 | (83%) | 12 | (92%) | 6 | (55%) | 0.05 | ||
| - Descending Thoracic Aorta, n (%) | 27 | (93%) | 10 | (77%) | 9 | (82%) | 0.21 | ||
| - Abdominal Aorta, n (%) | 26 | (90%) | 8 | (62%) | 8 | (73%) | 0.08 | ||
| - Other large vessel, n (%) | 20 | (69%) | 7 | (54%) | 6 | (55%) | 0.43 | ||
| No. of Sites affected | |||||||||
| - Single site, n (%) | 4 | (14%) | 1 | (8%) | 4 | (36%) | 0.14 | ||
| - ≥3 Sites, n (%) | 23 | (79%) | 8 | (62%) | 6 | (55%) | 0.24 | ||
| Treatment | 0.39 | ||||||||
| Standard Protocol, n (%) | 22 | (67%) | 13 | (81%) | 10 | (77%) | |||
| Steroids only, n (%) | 11 | (33%) | 3 | (19%) | 3 | (23%) | |||
| Outcomes | |||||||||
| Relapsing Patients, n (%) | 12 | (36%) | 8 | (50%) | 6 | (46%) | 0.62 | ||
| Refractory Disease, n (%) | 2 | (6%) | 5 | (31%) | 3 | (23%) | 0.05 | ||
Other traditional cardiovascular risk factors have long been associated with large vessel vasculitides, with some authors even postulating that GCA may in fact be a phenotype of cardiovascular disease [14]. Examining the prevalence of traditional cardiovascular risk factors in over 200 patients presenting with GCA, these authors identified a contributory effect of GCA to risk of a subsequent ischaemic event (OR 1.8; p=0.004). Of note however, the patients included were older (median 75 years) and no information on imaging or histology beyond the temporal artery was included.
Recent studies focusing on aortitis have acknowledged small numbers of SpAD patients within their cohorts [15-17]. Expanding upon our previous case series [7] and existing registry data [6], the current findings both confirm an association between LVV and SpAD-and establish some interesting nuances of LVV in these patients.
Although no significant differences in symptomatology or their duration were observed in our cohort, SpAD patients presented at a much younger age (59.2years vs. 70.3years iLVV and 68.1yrs AI; p=0.01). This de novo predisposition corroborates evidence from mouse models suggesting common pathophysiological traits between SpAD and large vessel inflammation [18]. Raised levels of Interleukin-23 (IL-23) contribute to both conditions, with IL-23 inoculated mice developing sacroiliitis and inflammation of the aortic root [18]. Treatment with systemic corticosteroids in both the autoimmune (AI) and SpAD groups did not influence age at LVV onset (p=0.58) nor the interval between symptom onset and diagnosis (p=0.79).
The relevance of seasonable symptom onset in identifying disease triggers has been subject to considerable debate [19, 20]. This concept however remains controversial, with others authors failing to identify any seasonal variation [21]. While SpAD patients tended to become symptomatic in spring (Fig. 2) the other groups exhibited only weak cyclical trends, again-supporting the concept that LVV in SpAD is phenotypically different and may be triggered by different pathogens or other environmental factors.
Of the serum inflammatory markers analysed, no differences were seen between SpAD patients and the other groups. Imaging however raised a number of interesting issues. Whilst positron emission tomography (PET-CT) has replaced magnetic resonance imaging (MRI) in treatment naïve SpAD patients with early disease [22, 23], it is nevertheless, important to stress that no inter-group bias relating to imaging modality existed (p=0.46). No difference in disease activity, signified by SUVmax, existed between groups suggesting a similar inflammatory intensity. Clear patterns of vessel involvement were identified, with a central preponderance in SpAD patients, particularly related to involvement of the ascending aorta (p=0.05) and sparing of the abdominal aorta (p=0.08) and large peripheral vessels, corroborating existing echocardiographic data associating aortic root inflammation with SpA [4, 5, 18].
Correspondingly, fewer SpAD patients demonstrated diffuse vessel involvement, although it should be noted that single-site disease was observed in only one patient. Considering these findings further, it would be reasonable to question if disease duration is confounding the results. However LVV onset in both AI and SpAD varied widely at median 21 [6 – 176] and 76 [16 – 201] months respectively (p=0.62). Considering both groups, duration of disease did not correlate with single-site (p=0.23) or diffuse LVV (p=0.54). Likewise, immunosuppression at or prior to LVV diagnosis did not influence single-site (p=0.77) or diffuse disease (p=0.63).
Regarding treatment response, while no differences in relapse rates between groups was observed, rates were highest among SpAD patients at 50%. Furthermore, these patients were at greater risk of developing recurrent relapses, with 31% being considered treatment refractory despite a wide-range of disease modifying treatments (p=0.05). Data relating to long-term outcomes in LVV is limited, with most reports suggesting no significant increases in mortality [24, 25].
Given its retrospective nature, a number of important limitations are inherent to the current analysis. The cohort’s small size cannot be ignored, especially given the long period over which patients were included. Given single centre recruitment, there is potential for inclusion bias. Treatment tapering was performed at discretion of the treating physician, introducing some variability in management particularly among patients without an additional inflammatory disease who were less likely to engage in long-term follow-up.
Collectively, these findings demonstrate an association between SpAD and LVV. Whilst clinically identical to that seen in other patient groups, it appears to occur at an earlier age, demonstrate seasonal variation and predominantly involve central vessels. Based on these findings, consideration of a coexisting SpAD should be encouraged in patients fitting this profile with no known past medical history. The aim of this study was to demonstrate an association between these two conditions. Our results reveal two distinct subsets of GCA, characterized by the absence or presence of SpAD. Recognition of this distinction may be important in interpreting treatment outcomes particularly where treatment responses are inconsistent, such as those seen with tocilizumab in SpA and LVV [26, 27]. Genome-wide association studies are currently evaluating susceptibility genes of GCA [28] and sub-analysis of these studies will ultimately reveal, whether or not the two subsets are clearly different.
LIST OF ABBREVIATIONS
| AI | = Autoimmune Disease |
| ASAS | = Assessment of Spondyloarthritis International Society |
| CT | = Computerized Tomography |
| DMARD | = Disease-Modifying Anti-Rheumatic Drugs |
| EULAR | = European League Against Rheumatism |
| GCA | = Giant-cell Arteritis |
| IL | = Interleukin |
| IQR | = Inter-quartile Range |
| LVV | = Large Vessel Vasculitis |
| MRI | = Magnetic Resonance Imaging |
| MTX | = Methotrexate |
| NSAID | = Non-Steroidal Anti-Inflammatory Drug |
| PET | = Positron-Emission Tomography |
| SpA | = Spondyloarthritis |
| SpAD | = Spondyloarthritis or its Associated Diseases |
| SUVmax | = Maximum Standard Uptake Value of 2-deoxy-2-18F-D-Glucose Labelled Tracer on Positron-Emission-Tomography |
| TA | = Takayasu’s Arteritis |
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared None.

